

Prion-Associated Neurodegeneration Causes Both Endoplasmic Reticulum Stress and Proteasome Impairment in a Murine Model of Spontaneous Disease
- PMID: 33466523
- PMCID: PMC7796520
- DOI: 10.3390/ijms22010465
Prion-Associated Neurodegeneration Causes Both Endoplasmic Reticulum Stress and Proteasome Impairment in a Murine Model of Spontaneous Disease
Abstract
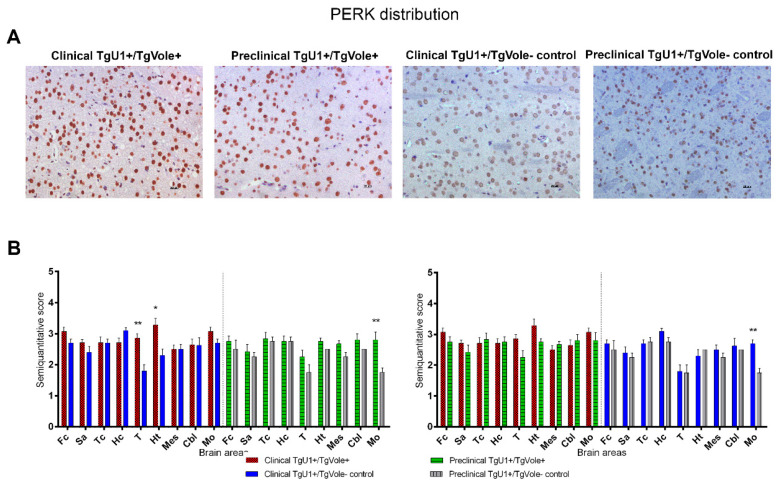
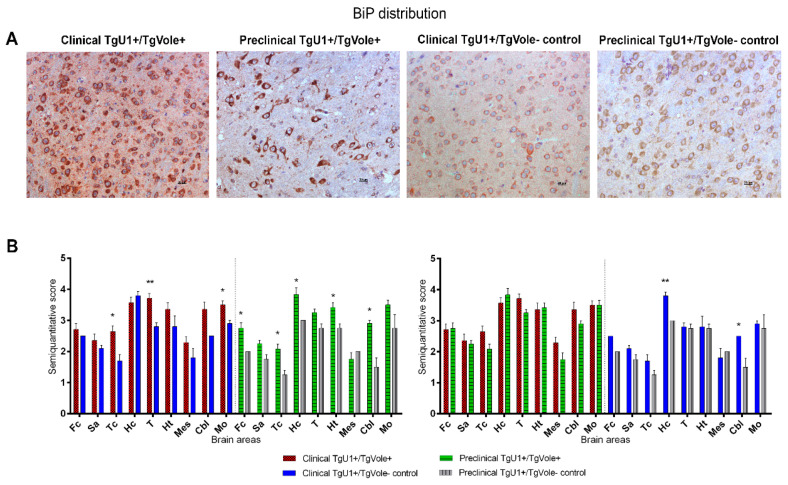
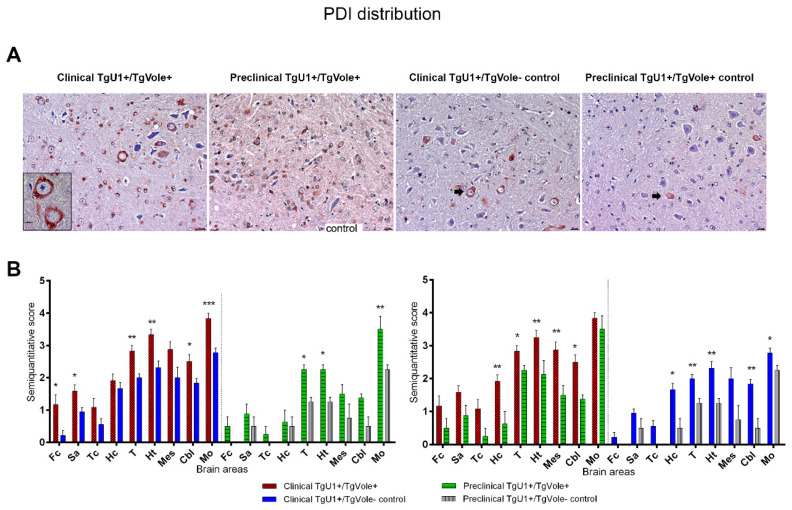
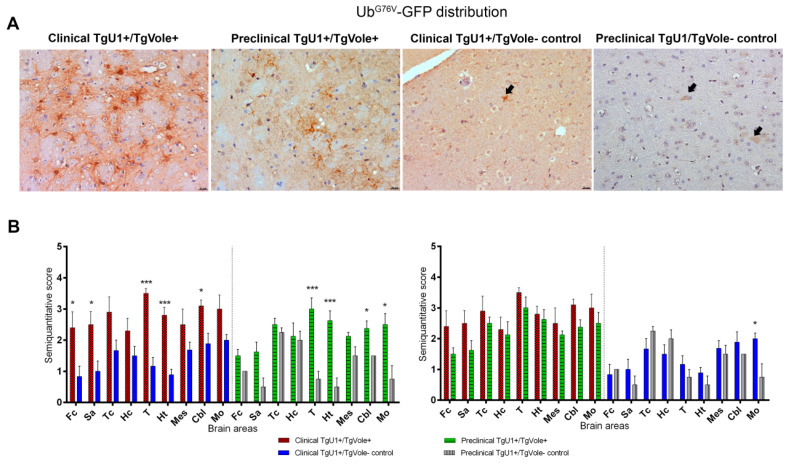
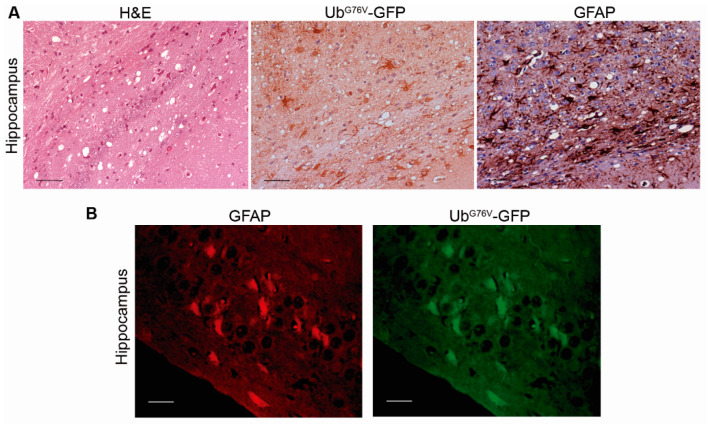
Prion diseases are a group of neurodegenerative disorders that can be spontaneous, familial or acquired by infection. The conversion of the prion protein PrPC to its abnormal and misfolded isoform PrPSc is the main event in the pathogenesis of prion diseases of all origins. In spontaneous prion diseases, the mechanisms that trigger the formation of PrPSc in the central nervous system remain unknown. Several reports have demonstrated that the accumulation of PrPSc can induce endoplasmic reticulum (ER) stress and proteasome impairment from the early stages of the prion disease. Both mechanisms lead to an increment of PrP aggregates in the secretory pathway, which could explain the pathogenesis of spontaneous prion diseases. Here, we investigate the role of ER stress and proteasome impairment during prion disorders in a murine model of spontaneous prion disease (TgVole) co-expressing the UbG76V-GFP reporter, which allows measuring the proteasome activity in vivo. Spontaneously prion-affected mice showed a significantly higher accumulation of the PKR-like ER kinase (PERK), the ER chaperone binding immunoglobulin protein (BiP/Grp78), the ER protein disulfide isomerase (PDI) and the UbG76V-GFP reporter than age-matched controls in certain brain areas. The upregulation of PERK, BiP, PDI and ubiquitin was detected from the preclinical stage of the disease, indicating that ER stress and proteasome impairment begin at early stages of the spontaneous disease. Strong correlations were found between the deposition of these markers and neuropathological markers of prion disease in both preclinical and clinical mice. Our results suggest that both ER stress and proteasome impairment occur during the pathogenesis of spontaneous prion diseases.
Keywords: ER stress; UPS impairment; endoplasmic reticulum; prions; proteasome.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
