

Identification of the Primary Factors Determining theSpecificity of Human VKORC1 Recognition by Thioredoxin-Fold Proteins
- PMID: 33466919
- PMCID: PMC7835823
- DOI: 10.3390/ijms22020802
Identification of the Primary Factors Determining theSpecificity of Human VKORC1 Recognition by Thioredoxin-Fold Proteins
Abstract
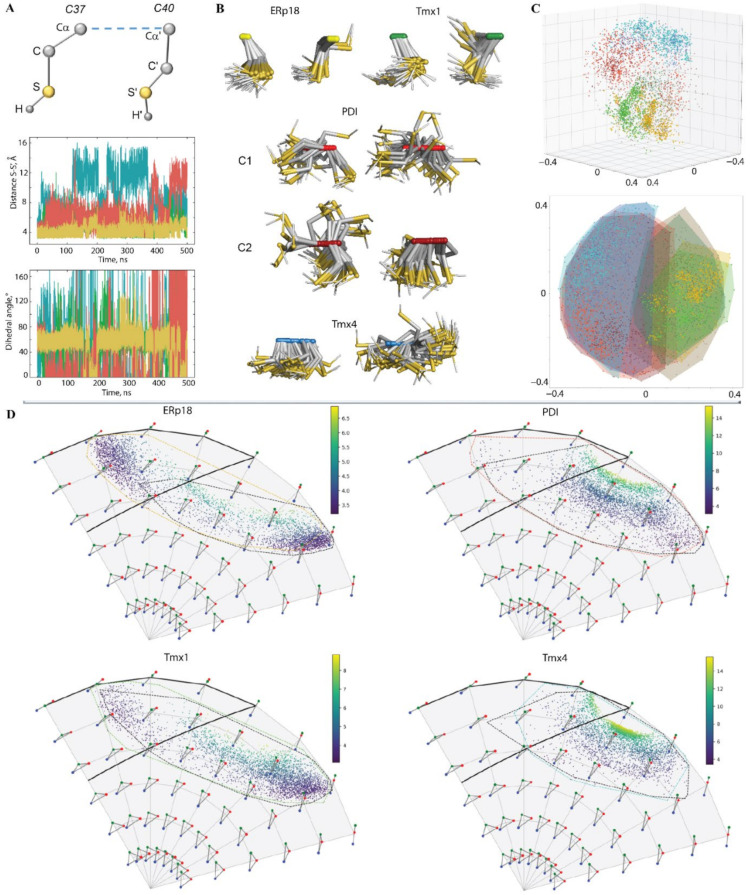
Redox (reduction-oxidation) reactions control many important biological processes in all organisms, both prokaryotes and eukaryotes. This reaction is usually accomplished by canonical disulphide-based pathways involving a donor enzyme that reduces the oxidised cysteine residues of a target protein, resulting in the cleavage of its disulphide bonds. Focusing on human vitamin K epoxide reductase (hVKORC1) as a target and on four redoxins (protein disulphide isomerase (PDI), endoplasmic reticulum oxidoreductase (ERp18), thioredoxin-related transmembrane protein 1 (Tmx1) and thioredoxin-related transmembrane protein 4 (Tmx4)) as the most probable reducers of VKORC1, a comparative in-silico analysis that concentrates on the similarity and divergence of redoxins in their sequence, secondary and tertiary structure, dynamics, intraprotein interactions and composition of the surface exposed to the target is provided. Similarly, hVKORC1 is analysed in its native state, where two pairs of cysteine residues are covalently linked, forming two disulphide bridges, as a target for Trx-fold proteins. Such analysis is used to derive the putative recognition/binding sites on each isolated protein, and PDI is suggested as the most probable hVKORC1 partner. By probing the alternative orientation of PDI with respect to hVKORC1, the functionally related noncovalent complex formed by hVKORC1 and PDI was found, which is proposed to be a first precursor to probe thiol-disulphide exchange reactions between PDI and hVKORC1.
Keywords: 3D modelling; PDI–hVKORC1 complex; Trx-fold proteins; dynamics; hVKORC1; molecular dynamics simulation; molecular recognition; protein folding; protein–protein interactions; thiol–disulphide exchange.
Conflict of interest statement
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Figures











Similar articles
-
Vitamin K Epoxide Reductase Complex-Protein Disulphide Isomerase Assemblies in the Thiol-Disulphide Exchange Reactions: Portrayal of Precursor-to-Successor Complexes.Int J Mol Sci. 2024 Apr 9;25(8):4135. doi: 10.3390/ijms25084135. Int J Mol Sci. 2024. PMID: 38673722 Free PMC article.
-
Human Vitamin K Epoxide Reductase as a Target of Its Redox Protein.Int J Mol Sci. 2022 Mar 31;23(7):3899. doi: 10.3390/ijms23073899. Int J Mol Sci. 2022. PMID: 35409257 Free PMC article.
-
Novel thioredoxin-related transmembrane protein TMX4 has reductase activity.J Biol Chem. 2010 Mar 5;285(10):7135-42. doi: 10.1074/jbc.M109.082545. Epub 2010 Jan 7. J Biol Chem. 2010. PMID: 20056998 Free PMC article.
-
The machinery for oxidative protein folding in thermophiles.Antioxid Redox Signal. 2008 Jan;10(1):157-69. doi: 10.1089/ars.2007.1855. Antioxid Redox Signal. 2008. PMID: 17956189 Review.
-
Enzymatic catalysis of disulfide formation.Protein Expr Purif. 1994 Feb;5(1):1-13. doi: 10.1006/prep.1994.1001. Protein Expr Purif. 1994. PMID: 7909462 Review.
Cited by
-
Introducing Thioredoxin-Related Transmembrane Proteins: Emerging Roles of Human TMX and Clinical Implications.Antioxid Redox Signal. 2022 May;36(13-15):984-1000. doi: 10.1089/ars.2021.0187. Epub 2021 Dec 7. Antioxid Redox Signal. 2022. PMID: 34465218 Free PMC article. Review.
-
Vitamin K Epoxide Reductase Complex-Protein Disulphide Isomerase Assemblies in the Thiol-Disulphide Exchange Reactions: Portrayal of Precursor-to-Successor Complexes.Int J Mol Sci. 2024 Apr 9;25(8):4135. doi: 10.3390/ijms25084135. Int J Mol Sci. 2024. PMID: 38673722 Free PMC article.
-
Human Vitamin K Epoxide Reductase as a Target of Its Redox Protein.Int J Mol Sci. 2022 Mar 31;23(7):3899. doi: 10.3390/ijms23073899. Int J Mol Sci. 2022. PMID: 35409257 Free PMC article.
-
Synergy of Mutation-Induced Effects in Human Vitamin K Epoxide Reductase: Perspectives and Challenges for Allo-Network Modulator Design.Int J Mol Sci. 2024 Feb 7;25(4):2043. doi: 10.3390/ijms25042043. Int J Mol Sci. 2024. PMID: 38396721 Free PMC article.
-
Special Issue: "Molecular Dynamics Simulations and Structural Analysis of Protein Domains".Int J Mol Sci. 2024 Oct 8;25(19):10793. doi: 10.3390/ijms251910793. Int J Mol Sci. 2024. PMID: 39409122 Free PMC article.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
