

Computational Functional Genomics-Based AmpliSeq™ Panel for Next-Generation Sequencing of Key Genes of Pain
- PMID: 33467215
- PMCID: PMC7830224
- DOI: 10.3390/ijms22020878
Computational Functional Genomics-Based AmpliSeq™ Panel for Next-Generation Sequencing of Key Genes of Pain
Abstract
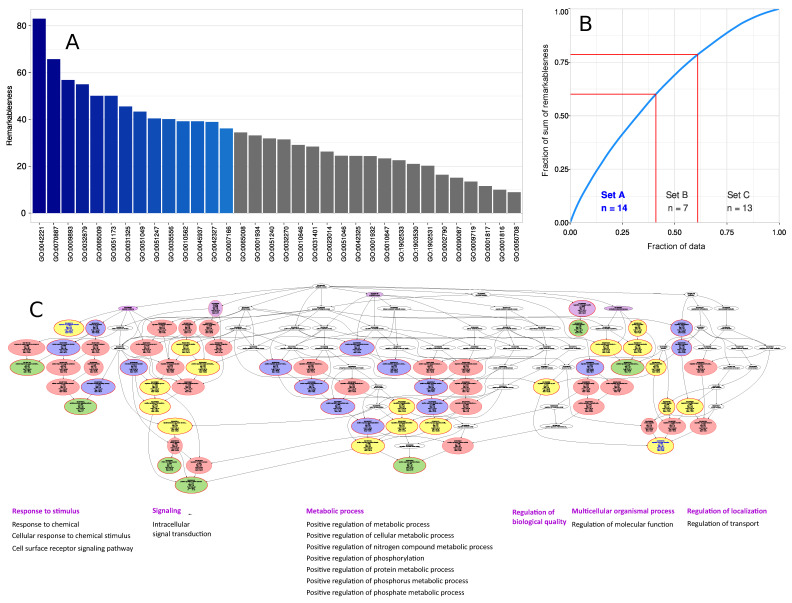
The genetic background of pain is becoming increasingly well understood, which opens up possibilities for predicting the individual risk of persistent pain and the use of tailored therapies adapted to the variant pattern of the patient's pain-relevant genes. The individual variant pattern of pain-relevant genes is accessible via next-generation sequencing, although the analysis of all "pain genes" would be expensive. Here, we report on the development of a cost-effective next generation sequencing-based pain-genotyping assay comprising the development of a customized AmpliSeq™ panel and bioinformatics approaches that condensate the genetic information of pain by identifying the most representative genes. The panel includes 29 key genes that have been shown to cover 70% of the biological functions exerted by a list of 540 so-called "pain genes" derived from transgenic mice experiments. These were supplemented by 43 additional genes that had been independently proposed as relevant for persistent pain. The functional genomics covered by the resulting 72 genes is particularly represented by mitogen-activated protein kinase of extracellular signal-regulated kinase and cytokine production and secretion. The present genotyping assay was established in 61 subjects of Caucasian ethnicity and investigates the functional role of the selected genes in the context of the known genetic architecture of pain without seeking functional associations for pain. The assay identified a total of 691 genetic variants, of which many have reports for a clinical relevance for pain or in another context. The assay is applicable for small to large-scale experimental setups at contemporary genotyping costs.
Keywords: computational functional genomics; data science; human genomics; knowledge discovery; next generation sequencing; pain genetics; pharmacogenomics.
Conflict of interest statement
The authors have declared that no further conflict of interest exist.
Figures




Similar articles
-
Development of an AmpliSeqTM Panel for Next-Generation Sequencing of a Set of Genetic Predictors of Persisting Pain.Front Pharmacol. 2018 Sep 19;9:1008. doi: 10.3389/fphar.2018.01008. eCollection 2018. Front Pharmacol. 2018. PMID: 30283335 Free PMC article.
-
Inter-laboratory evaluation of SNP-based forensic identification by massively parallel sequencing using the Ion PGM™.Forensic Sci Int Genet. 2015 Jul;17:110-121. doi: 10.1016/j.fsigen.2015.04.007. Epub 2015 Apr 15. Forensic Sci Int Genet. 2015. PMID: 25955683
-
Integration of Technical, Bioinformatic, and Variant Assessment Approaches in the Validation of a Targeted Next-Generation Sequencing Panel for Myeloid Malignancies.Arch Pathol Lab Med. 2017 Jun;141(6):759-775. doi: 10.5858/arpa.2016-0547-RA. Epub 2017 Mar 9. Arch Pathol Lab Med. 2017. PMID: 28557600
-
Requirements for comprehensive pharmacogenetic genotyping platforms.Pharmacogenomics. 2016 Jun;17(8):917-24. doi: 10.2217/pgs-2016-0023. Epub 2016 Jun 1. Pharmacogenomics. 2016. PMID: 27248710 Review.
-
Critical review of NGS analyses for de novo genotyping multigene families.Mol Ecol. 2014 Aug;23(16):3957-72. doi: 10.1111/mec.12843. Epub 2014 Jul 21. Mol Ecol. 2014. PMID: 24954669 Review.
Cited by
-
Pharmacokinetic and neuroimmune pharmacogenetic impacts on slow-release morphine cancer pain control and adverse effects.Pharmacogenomics J. 2024 Jun 1;24(3):18. doi: 10.1038/s41397-024-00339-w. Pharmacogenomics J. 2024. PMID: 38824169 Free PMC article.
References
-
- Navratilova E., Rau J., Oyarzo J., Tien J., Mackenzie K., Stratton J., Remeniuk B., Schwedt T., Anderson T., Dodick D., et al. CGRP-dependent and independent mechanisms of acute and persistent post-traumatic headache following mild traumatic brain injury in mice. Cephalalgia. 2019;39:1762–1775. doi: 10.1177/0333102419877662. - DOI - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
