

IFN-γ and CD38 in Hyperprogressive Cancer Development
- PMID: 33467713
- PMCID: PMC7830527
- DOI: 10.3390/cancers13020309
IFN-γ and CD38 in Hyperprogressive Cancer Development
Abstract
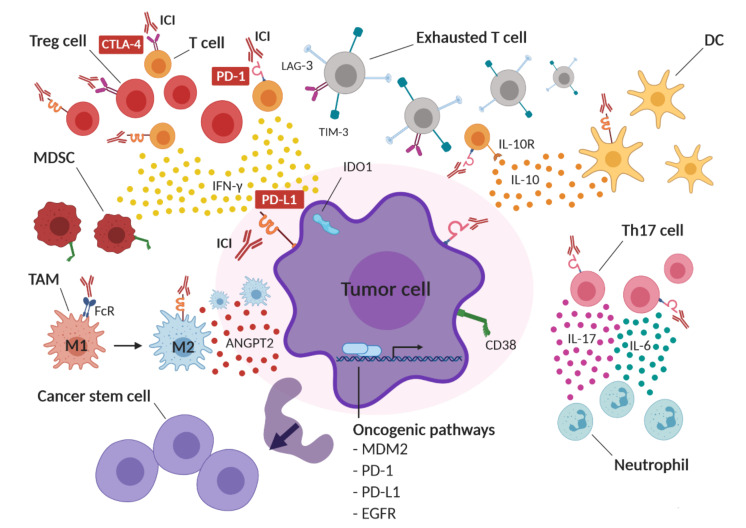
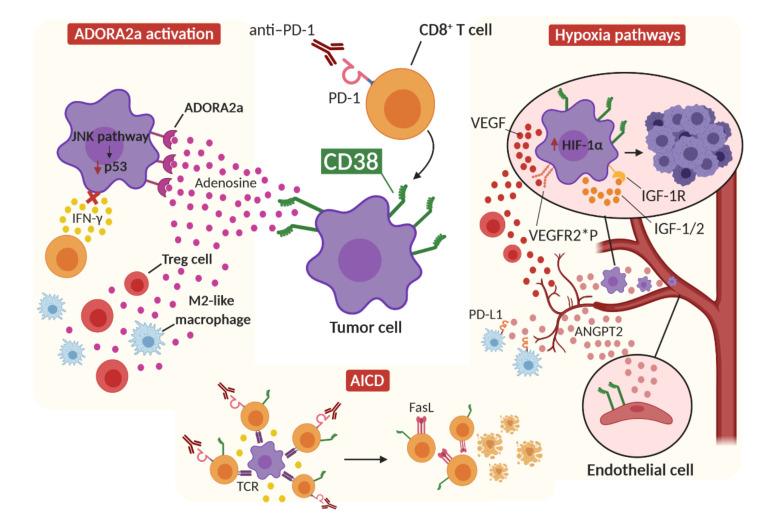
Immune checkpoint inhibitors (ICIs) improve the survival of patients with multiple types of cancer. However, low response rates and atypical responses limit their success in clinical applications. The paradoxical acceleration of tumor growth after treatment, defined as hyperprogressive disease (HPD), is the most difficult problem facing clinicians and patients alike. The mechanisms that underlie hyperprogression (HP) are still unclear and controversial, although different factors are associated with the phenomenon. In this review, we propose two factors that have not yet been demonstrated to be directly associated with HP, but upon which it is important to focus attention. IFN-γ is a key cytokine in antitumor response and its levels increase during ICI therapy, whereas CD38 is an alternative immune checkpoint that is involved in immunosuppressive responses. As both factors are associated with resistance to ICI therapy, we have discussed their possible involvement in HPD with the conclusion that IFN-γ may contribute to HP onset through the activation of the inflammasome pathway, immunosuppressive enzyme IDO1 and activation-induced cell death (AICD) in effector T cells, while the role of CD38 in HP may be associated with the activation of adenosine receptors, hypoxia pathways and AICD-dependent T-cell depletion.
Keywords: CD38; IFN-γ; cancer; hyperprogression; hyperprogressive disease; immune checkpoint inhibitors; immunotherapy; macrophage; tumor microenvironment.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

References
-
- Topalian S.L., Sznol M., McDermott D.F., Kluger H.M., Carvajal R.D., Sharfman W.H., Brahmer J.R., Lawrence D.P., Atkins M.B., Powderly J.D., et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J. Clin. Oncol. 2014;32:1020–1030. doi: 10.1200/JCO.2013.53.0105. - DOI - PMC - PubMed
-
- Ahamadi M., Freshwater T., Prohn M., Li C., de Alwis D., de Greef R., Elassaiss-Schaap J., Kondic A., Stone J. Model-Based Characterization of the Pharmacokinetics of Pembrolizumab: A Humanized Anti-PD-1 Monoclonal Antibody in Advanced Solid Tumors. CPT Pharmacomet. Syst. Pharmacol. 2017;6:49–57. doi: 10.1002/psp4.12139. - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
