

Deletion of pknG Abates Reactivation of Latent Mycobacterium tuberculosis in Mice
- PMID: 33468473
- PMCID: PMC8097433
- DOI: 10.1128/AAC.02095-20
Deletion of pknG Abates Reactivation of Latent Mycobacterium tuberculosis in Mice
Abstract
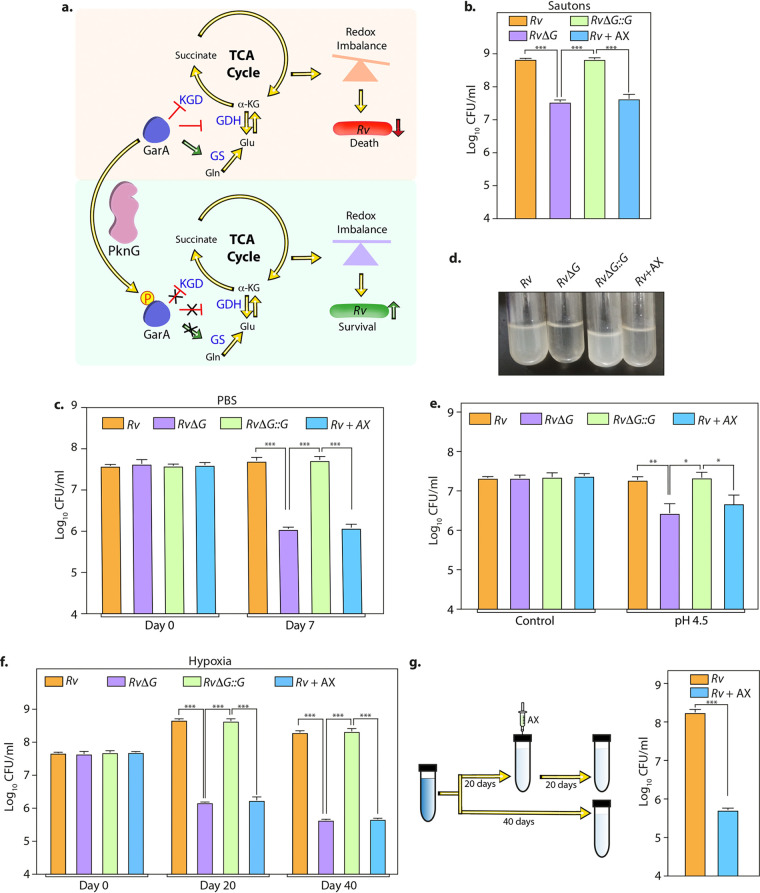
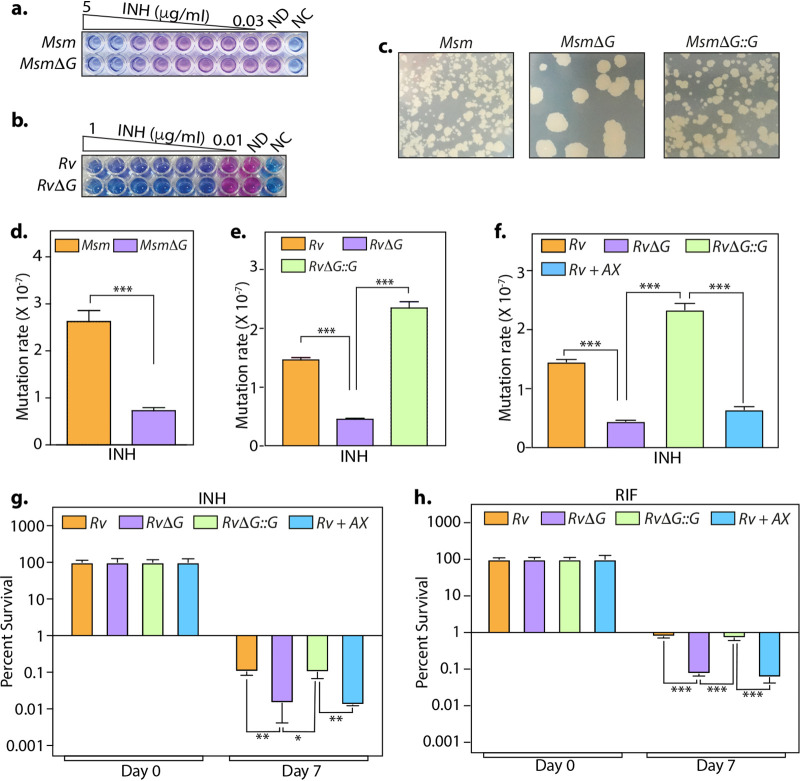
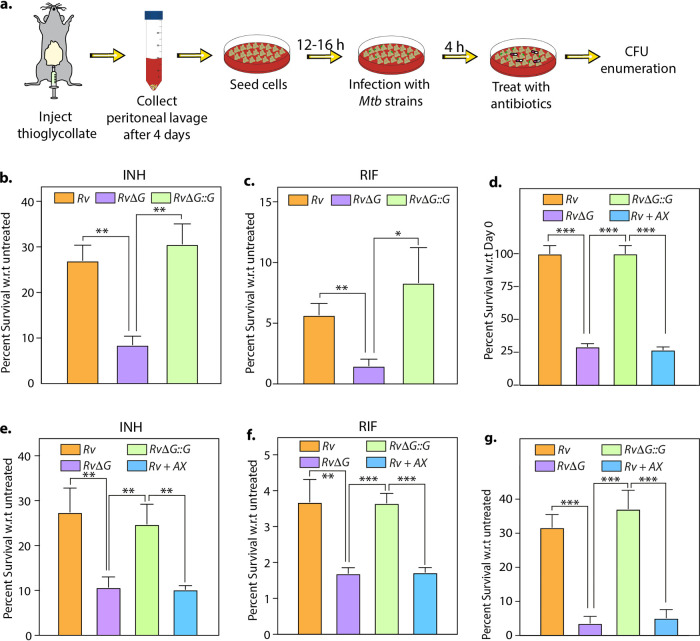
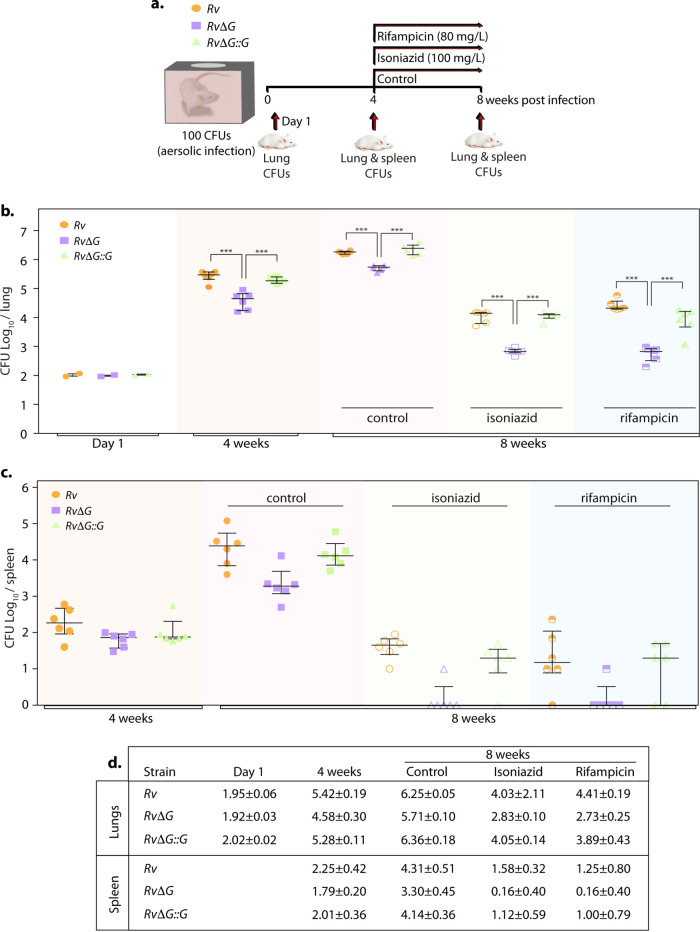
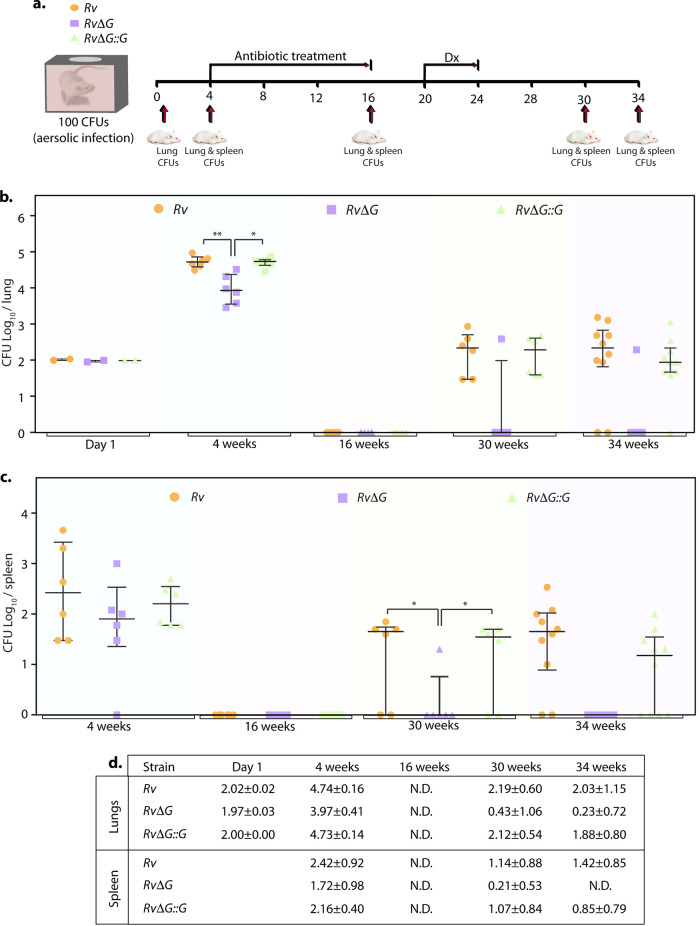
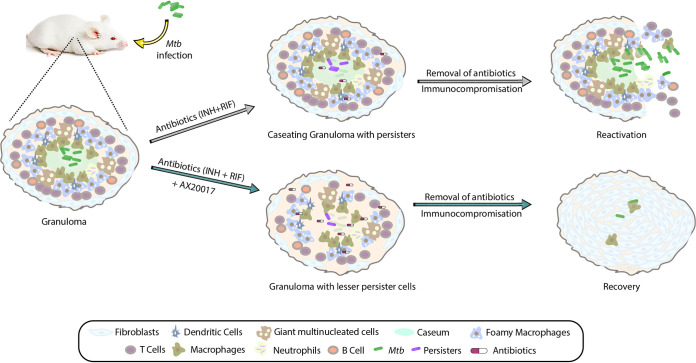
Eradication of tuberculosis (TB), caused by Mycobacterium tuberculosis (Mtb), has been a challenge due to its uncanny ability to survive in a dormant state inside host granulomas for decades. Mtb rewires its metabolic and redox regulatory networks to survive in the hostile hypoxic and nutrient-limiting environment, facilitating the formation of drug-tolerant persisters. Previously, we showed that protein kinase G (PknG), a virulence factor required for lysosomal escape, aids in metabolic adaptation, thereby promoting the survival of nonreplicating mycobacteria. Here, we sought to investigate the therapeutic potential of PknG against latent mycobacterium. We show that inhibition of PknG by AX20017 reduces mycobacterial survival in in vitro latency models such as hypoxia, persisters, and nutrient starvation. Targeting PknG enhances the bactericidal activity of the frontline anti-TB drugs in peritoneal macrophages. Deletion of pknG resulted in 5- to 15-fold-reduced survival of Mtb in chronically infected mice treated with anti-TB drugs. Importantly, in the Cornell mouse model of latent TB, the deletion of pknG drastically attenuated Mtb's ability to resuscitate after antibiotic treatment compared with wild-type and complemented strains. This is the first study to investigate the sterilizing activity of pknG deletion and inhibition for adjunct therapy against latent TB in a preclinical model. Collectively, these results suggest that PknG may be a promising drug target for adjunct therapy to shorten the treatment duration and reduce disease relapse.
Keywords: Mycobacterium tuberculosis; adjunct therapy; bacterial protein kinase; bacterial signal transduction; drug discovery; hypoxia; latency; latent infection; persistence; persisters; phosphorylation.
Copyright © 2021 American Society for Microbiology.
Figures
Similar articles
-
Protein kinase G confers survival advantage to Mycobacterium tuberculosis during latency-like conditions.J Biol Chem. 2017 Sep 29;292(39):16093-16108. doi: 10.1074/jbc.M117.797563. Epub 2017 Aug 18. J Biol Chem. 2017. PMID: 28821621 Free PMC article.
-
Sclerotiorin inhibits protein kinase G from Mycobacterium tuberculosis and impairs mycobacterial growth in macrophages.Tuberculosis (Edinb). 2017 Mar;103:37-43. doi: 10.1016/j.tube.2017.01.001. Epub 2017 Jan 12. Tuberculosis (Edinb). 2017. PMID: 28237032
-
[Recent progress in mycobacteriology].Kekkaku. 2007 Oct;82(10):783-99. Kekkaku. 2007. PMID: 18018602 Japanese.
-
The significance of persisters in tuberculosis drug discovery: Exploring the potential of targeting the glyoxylate shunt pathway.Eur J Med Chem. 2024 Feb 5;265:116058. doi: 10.1016/j.ejmech.2023.116058. Epub 2023 Dec 18. Eur J Med Chem. 2024. PMID: 38128237 Review.
-
Drug targets in dormant Mycobacterium tuberculosis: can the conquest against tuberculosis become a reality?Infect Dis (Lond). 2018 Feb;50(2):81-94. doi: 10.1080/23744235.2017.1377346. Epub 2017 Sep 21. Infect Dis (Lond). 2018. PMID: 28933243 Review.
Cited by
-
Glutamate decarboxylase confers acid tolerance and enhances survival of mycobacteria within macrophages.J Biol Chem. 2025 Apr;301(4):108338. doi: 10.1016/j.jbc.2025.108338. Epub 2025 Feb 21. J Biol Chem. 2025. PMID: 39988078 Free PMC article.
-
Mycobacterium tuberculosis and its clever approaches to escape the deadly macrophage.World J Microbiol Biotechnol. 2023 Sep 5;39(11):300. doi: 10.1007/s11274-023-03735-9. World J Microbiol Biotechnol. 2023. PMID: 37667129 Review.
-
Divergent downstream biosynthetic pathways are supported by <sc>L</sc>-cysteine synthases of Mycobacterium tuberculosis.Elife. 2024 Aug 29;12:RP91970. doi: 10.7554/eLife.91970. Elife. 2024. PMID: 39207917 Free PMC article.
-
Pathogenicity and virulence of Mycobacterium tuberculosis.Virulence. 2023 Dec;14(1):2150449. doi: 10.1080/21505594.2022.2150449. Virulence. 2023. PMID: 36419223 Free PMC article. Review.
-
Characterization of the Biological Effect Mediated by Mycobacterial Kinase PknG on Protein Phosphorylation and Acetylation.ACS Omega. 2025 May 21;10(21):21662-21673. doi: 10.1021/acsomega.5c01032. eCollection 2025 Jun 3. ACS Omega. 2025. PMID: 40488057 Free PMC article.
References
-
- Getahun H, Matteelli A, Abubakar I, Aziz MA, Baddeley A, Barreira D, Den Boon S, Borroto Gutierrez SM, Bruchfeld J, Burhan E, Cavalcante S, Cedillos R, Chaisson R, Chee CB-E, Chesire L, Corbett E, Dara M, Denholm J, de Vries G, Falzon D, Ford N, Gale-Rowe M, Gilpin C, Girardi E, Go U-Y, Govindasamy D, Grzemska M, Harris R, Horsburgh CR, Jr, Ismayilov A, Jaramillo E, Kik S, Kranzer K, Lienhardt C, LoBue P, Lonnroth K, Marks G, Menzies D, Migliori GB, Mosca D, Mukadi YD, Mwinga A, Nelson L, Nishikiori N, Oordt-Speets A, Rangaka MX, Reis A, Rotz L, Sandgren A, Sane Schepisi M, et al.. 2015. Management of latent Mycobacterium tuberculosis infection: WHO guidelines for low tuberculosis burden countries. Eur Respir J 46:1563–1576. doi:10.1183/13993003.01245-2015. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
