

Disease Modeling with Human Neurons Reveals LMNB1 Dysregulation Underlying DYT1 Dystonia
- PMID: 33468570
- PMCID: PMC7939088
- DOI: 10.1523/JNEUROSCI.2507-20.2020
Disease Modeling with Human Neurons Reveals LMNB1 Dysregulation Underlying DYT1 Dystonia
Abstract
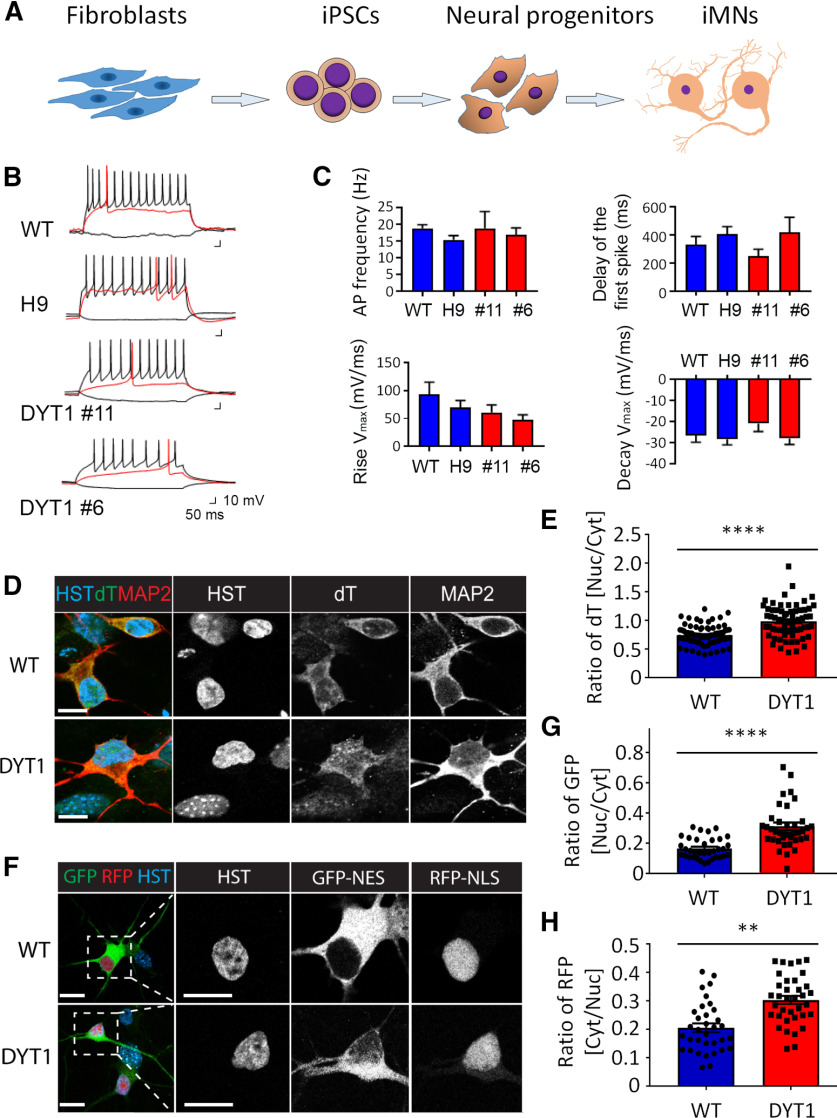
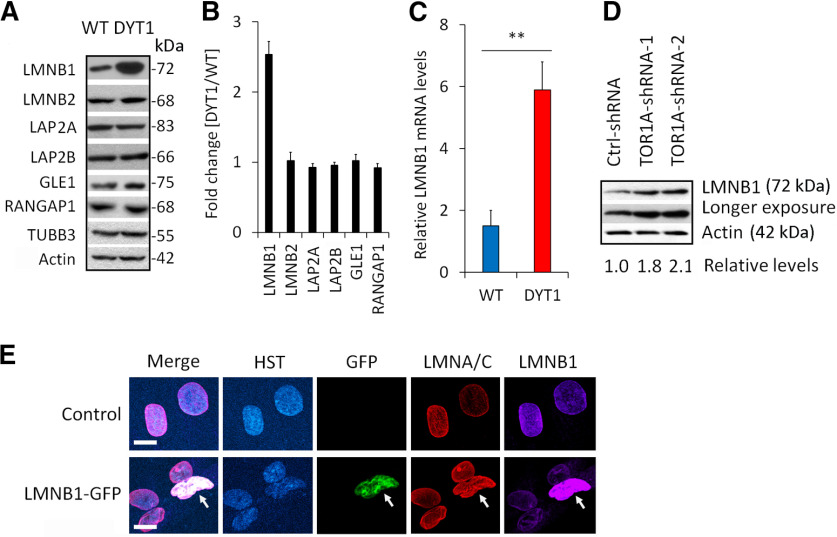
DYT1 dystonia is a hereditary neurologic movement disorder characterized by uncontrollable muscle contractions. It is caused by a heterozygous mutation in Torsin A (TOR1A), a gene encoding a membrane-embedded ATPase. While animal models provide insights into disease mechanisms, significant species-dependent differences exist since animals with the identical heterozygous mutation fail to show pathology. Here, we model DYT1 by using human patient-specific cholinergic motor neurons (MNs) that are generated through either direct conversion of patients' skin fibroblasts or differentiation of induced pluripotent stem cells (iPSCs). These human MNs with the heterozygous TOR1A mutation show reduced neurite length and branches, markedly thickened nuclear lamina, disrupted nuclear morphology, and impaired nucleocytoplasmic transport (NCT) of mRNAs and proteins, whereas they lack the perinuclear "blebs" that are often observed in animal models. Furthermore, we uncover that the nuclear lamina protein LMNB1 is upregulated in DYT1 cells and exhibits abnormal subcellular distribution in a cholinergic MNs-specific manner. Such dysregulation of LMNB1 can be recapitulated by either ectopic expression of the mutant TOR1A gene or shRNA-mediated downregulation of endogenous TOR1A in healthy control MNs. Interestingly, downregulation of LMNB1 can largely ameliorate all the cellular defects in DYT1 MNs. These results reveal the value of disease modeling with human patient-specific neurons and indicate that dysregulation of LMNB1, a crucial component of the nuclear lamina, may constitute a major molecular mechanism underlying DYT1 pathology.SIGNIFICANCE STATEMENT Inaccessibility to patient neurons greatly impedes our understanding of the pathologic mechanisms for dystonia. In this study, we employ reprogrammed human patient-specific motor neurons (MNs) to model DYT1, the most severe hereditary form of dystonia. Our results reveal disease-dependent deficits in nuclear morphology and nucleocytoplasmic transport (NCT). Most importantly, we further identify LMNB1 dysregulation as a major contributor to these deficits, uncovering a new pathologic mechanism for DYT1 dystonia.
Keywords: TOR1A; cholinergic motor neurons; dystonia; human neurons; nuclear LMNB1; nucleocytoplasmic transport.
Copyright © 2021 the authors.
Figures







Similar articles
-
RANBP17 Overexpression Restores Nucleocytoplasmic Transport and Ameliorates Neurodevelopment in Induced DYT1 Dystonia Motor Neurons.J Neurosci. 2024 Apr 10;44(15):e1728232024. doi: 10.1523/JNEUROSCI.1728-23.2024. J Neurosci. 2024. PMID: 38438257 Free PMC article.
-
Decreased number of striatal cholinergic interneurons and motor deficits in dopamine receptor 2-expressing-cell-specific Dyt1 conditional knockout mice.Neurobiol Dis. 2020 Feb;134:104638. doi: 10.1016/j.nbd.2019.104638. Epub 2019 Oct 13. Neurobiol Dis. 2020. PMID: 31618684 Free PMC article.
-
Cell-specific effects of Dyt1 knock-out on sensory processing, network-level connectivity, and motor deficits.Exp Neurol. 2021 Sep;343:113783. doi: 10.1016/j.expneurol.2021.113783. Epub 2021 Jun 10. Exp Neurol. 2021. PMID: 34119482 Free PMC article.
-
Membrane defects and genetic redundancy: Are we at a turning point for DYT1 dystonia?Mov Disord. 2017 Mar;32(3):371-381. doi: 10.1002/mds.26880. Epub 2016 Dec 2. Mov Disord. 2017. PMID: 27911022 Review.
-
Non-DYT1 early-onset primary torsion dystonia: comparison with DYT1 phenotype and review of the literature.Mov Disord. 2006 Sep;21(9):1411-8. doi: 10.1002/mds.21000. Mov Disord. 2006. PMID: 16773641 Review.
Cited by
-
Atypical nuclear envelope condensates linked to neurological disorders reveal nucleoporin-directed chaperone activities.Nat Cell Biol. 2022 Nov;24(11):1630-1641. doi: 10.1038/s41556-022-01001-y. Epub 2022 Oct 27. Nat Cell Biol. 2022. PMID: 36302970 Free PMC article.
-
Rapid and high-purity differentiation of human medium spiny neurons reveals LMNB1 hypofunction and subtype necessity in modeling Huntington's disease.Inflamm Regen. 2024 Feb 15;44(1):7. doi: 10.1186/s41232-024-00320-x. Inflamm Regen. 2024. PMID: 38360694 Free PMC article.
-
Novel insights into the pathogenesis of DYT1 dystonia from induced patient-derived neurons.Neural Regen Res. 2022 Mar;17(3):561-562. doi: 10.4103/1673-5374.320978. Neural Regen Res. 2022. PMID: 34380890 Free PMC article. No abstract available.
-
Modeling Movement Disorders via Generation of hiPSC-Derived Motor Neurons.Cells. 2022 Nov 27;11(23):3796. doi: 10.3390/cells11233796. Cells. 2022. PMID: 36497056 Free PMC article. Review.
-
Screens in aging-relevant human ALS-motor neurons identify MAP4Ks as therapeutic targets for the disease.Cell Death Dis. 2024 Jan 4;15(1):4. doi: 10.1038/s41419-023-06395-7. Cell Death Dis. 2024. PMID: 38177100 Free PMC article.
References
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials