

The Tiger Rattlesnake genome reveals a complex genotype underlying a simple venom phenotype
- PMID: 33468678
- PMCID: PMC7848695
- DOI: 10.1073/pnas.2014634118
The Tiger Rattlesnake genome reveals a complex genotype underlying a simple venom phenotype
Abstract
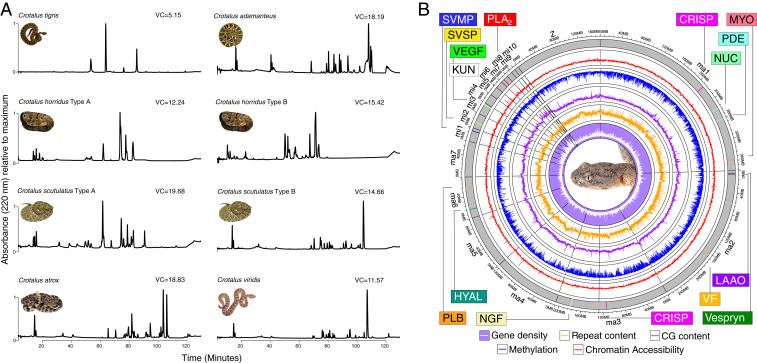
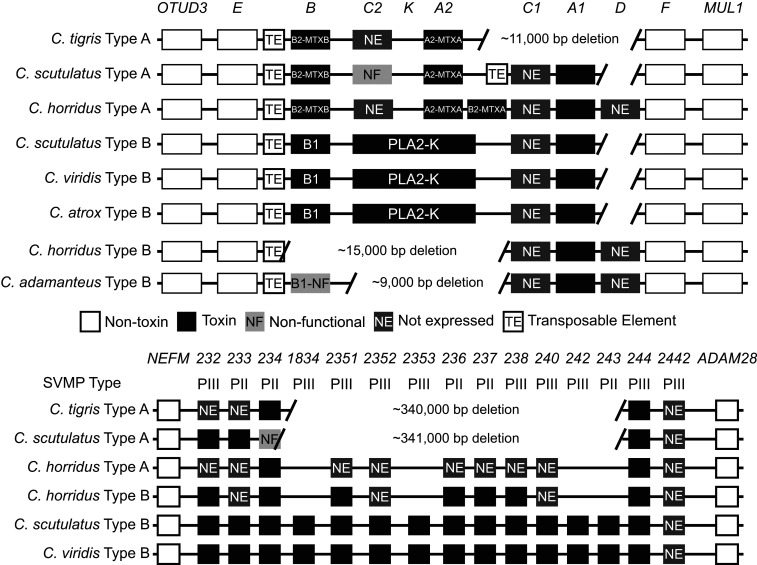
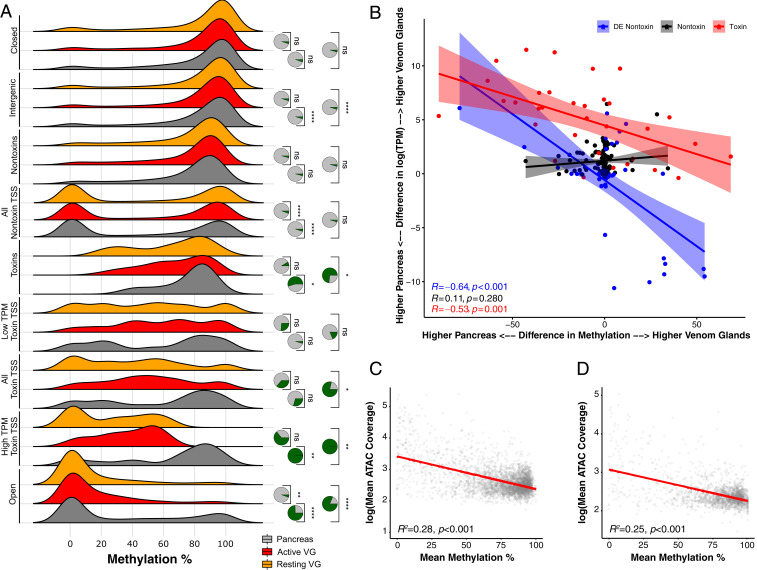
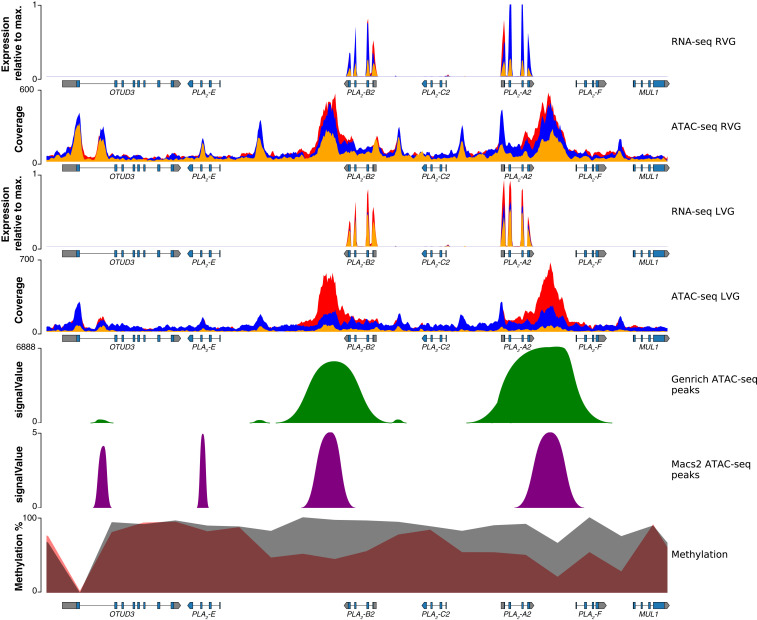
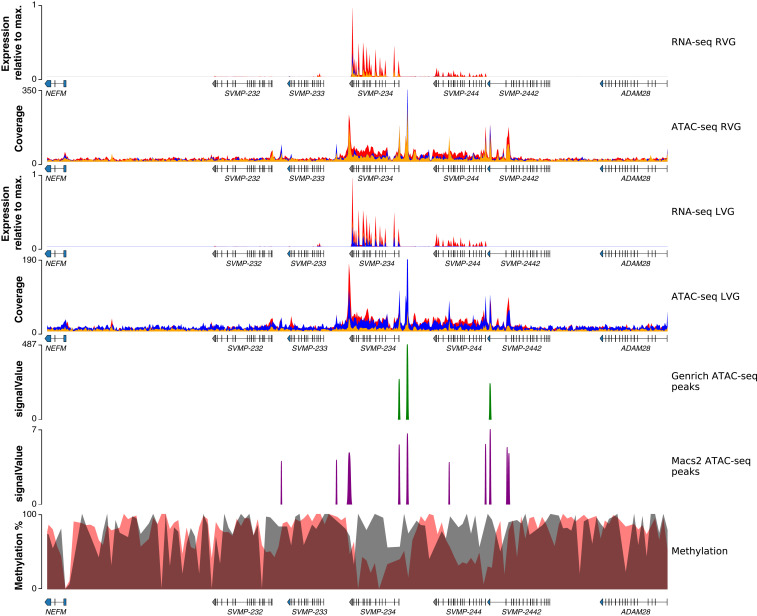
Variation in gene regulation is ubiquitous, yet identifying the mechanisms producing such variation, especially for complex traits, is challenging. Snake venoms provide a model system for studying the phenotypic impacts of regulatory variation in complex traits because of their genetic tractability. Here, we sequence the genome of the Tiger Rattlesnake, which possesses the simplest and most toxic venom of any rattlesnake species, to determine whether the simple venom phenotype is the result of a simple genotype through gene loss or a complex genotype mediated through regulatory mechanisms. We generate the most contiguous snake-genome assembly to date and use this genome to show that gene loss, chromatin accessibility, and methylation levels all contribute to the production of the simplest, most toxic rattlesnake venom. We provide the most complete characterization of the venom gene-regulatory network to date and identify key mechanisms mediating phenotypic variation across a polygenic regulatory network.
Keywords: chromatin; gene regulation; genotype–phenotype; methylation.
Conflict of interest statement
The authors declare no competing interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
