

DNA methylation perturbations may link altered development and aging in the lung
- PMID: 33468710
- PMCID: PMC7880367
- DOI: 10.18632/aging.202544
DNA methylation perturbations may link altered development and aging in the lung
Abstract
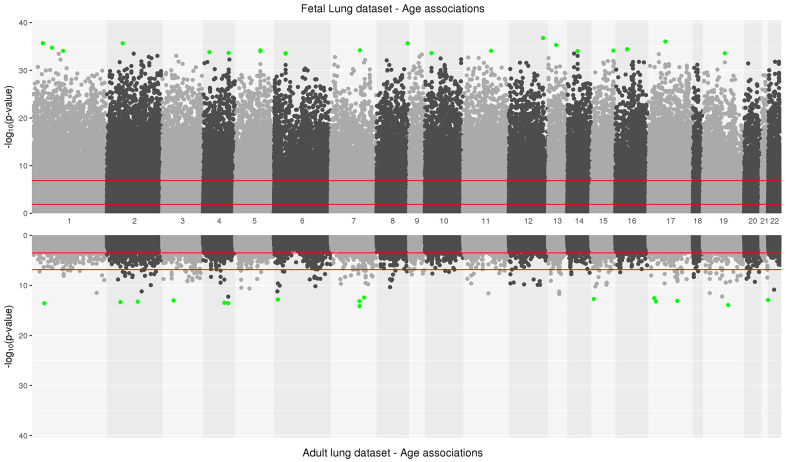
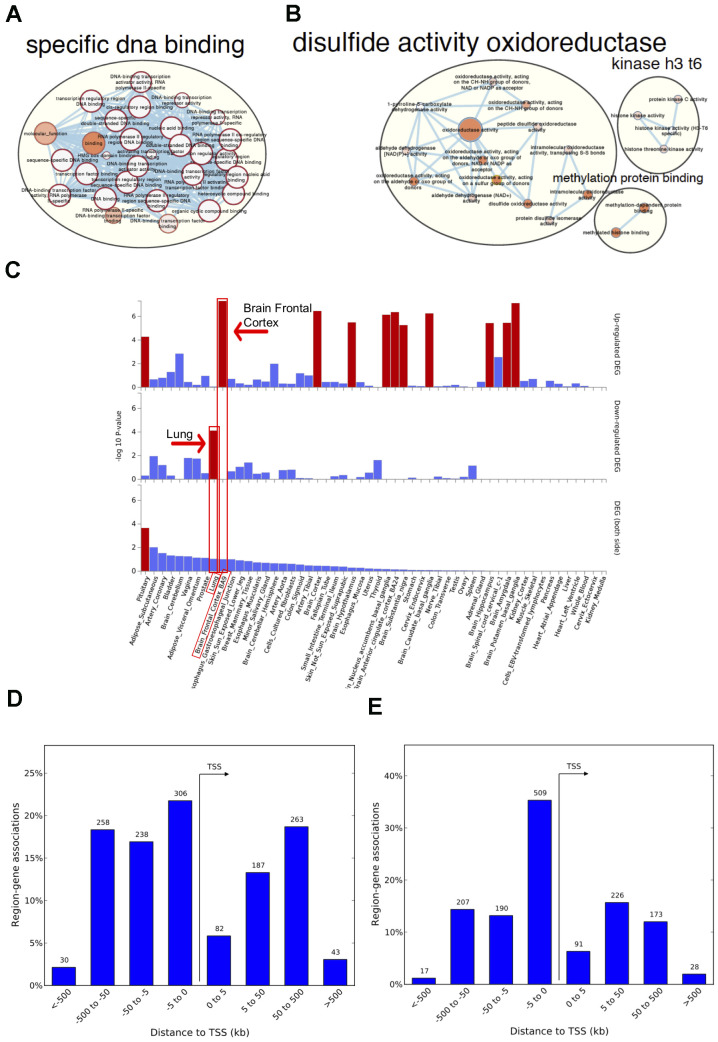
Fetal perturbations in DNA methylation during lung development may reveal insights into the enduring impacts on adult lung health and disease during aging that have not been explored altogether before. We studied the association between genome-wide DNA-methylation and post-conception age in fetal-lung (n=78, 42 exposed to in-utero-smoke (IUS)) tissue and chronological age in adult-lung tissue (n=160, 114 with Chronic Obstructive Pulmonary Disease) using multi-variate linear regression models with covariate adjustment and tested for effect modification by phenotypes. Overlapping age-associations were evaluated for functional and tissue-specific enrichment using the Genotype-Tissue-Expression (GTEx) project. We identified 244 age-associated differentially methylated positions and 878 regions overlapping between fetal and adult-lung tissues. Hyper-methylated CpGs (96%) were enriched in transcription factor activity (FDR adjusted P=2x10-33) and implicated in developmental processes including embryonic organ morphogenesis, neurogenesis and growth delay. Hypo-methylated CpGs (2%) were enriched in oxido-reductase activity and VEGFA-VEGFR2 Signaling. Twenty-one age-by-sex and eleven age-by-pack-years interactions were statistically significant (FDR<0.05) in adult-lung tissue. DNA methylation in transcription factors during development in fetal lung recapitulates in adult-lung tissue with aging. These findings reveal molecular mechanisms and pathways that may link disrupted development in early-life and age-associated lung diseases.
Keywords: DNA methylation; aging; development; lung; transcription factors.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
