

Interplay between cofactors and transcription factors in hematopoiesis and hematological malignancies
- PMID: 33468999
- PMCID: PMC7815747
- DOI: 10.1038/s41392-020-00422-1
Interplay between cofactors and transcription factors in hematopoiesis and hematological malignancies
Abstract
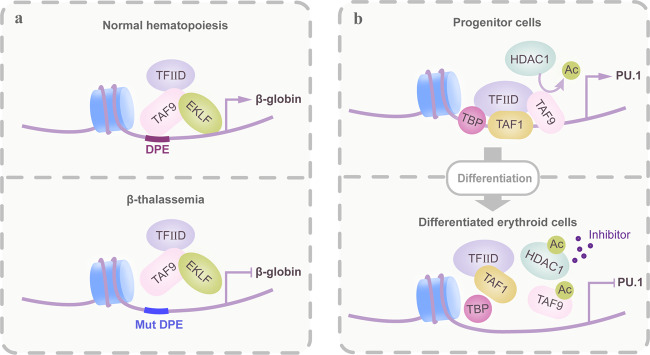
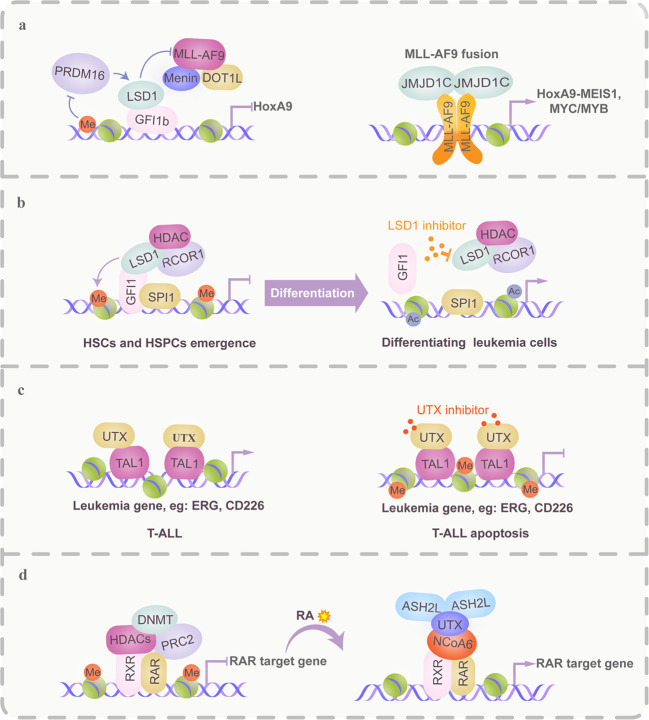
Hematopoiesis requires finely tuned regulation of gene expression at each stage of development. The regulation of gene transcription involves not only individual transcription factors (TFs) but also transcription complexes (TCs) composed of transcription factor(s) and multisubunit cofactors. In their normal compositions, TCs orchestrate lineage-specific patterns of gene expression and ensure the production of the correct proportions of individual cell lineages during hematopoiesis. The integration of posttranslational and conformational modifications in the chromatin landscape, nucleosomes, histones and interacting components via the cofactor-TF interplay is critical to optimal TF activity. Mutations or translocations of cofactor genes are expected to alter cofactor-TF interactions, which may be causative for the pathogenesis of various hematologic disorders. Blocking TF oncogenic activity in hematologic disorders through targeting cofactors in aberrant complexes has been an exciting therapeutic strategy. In this review, we summarize the current knowledge regarding the models and functions of cofactor-TF interplay in physiological hematopoiesis and highlight their implications in the etiology of hematological malignancies. This review presents a deep insight into the physiological and pathological implications of transcription machinery in the blood system.
Conflict of interest statement
The authors declare no competing interests.
Figures




References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
