

Targeting cancer-promoting inflammation - have anti-inflammatory therapies come of age?
- PMID: 33469195
- PMCID: PMC8978805
- DOI: 10.1038/s41571-020-00459-9
Targeting cancer-promoting inflammation - have anti-inflammatory therapies come of age?
Abstract
The immune system has crucial roles in cancer development and treatment. Whereas adaptive immunity can prevent or constrain cancer through immunosurveillance, innate immunity and inflammation often promote tumorigenesis and malignant progression of nascent cancer. The past decade has witnessed the translation of knowledge derived from preclinical studies of antitumour immunity into clinically effective, approved immunotherapies for cancer. By contrast, the successful implementation of treatments that target cancer-associated inflammation is still awaited. Anti-inflammatory agents have the potential to not only prevent or delay cancer onset but also to improve the efficacy of conventional therapeutics and next-generation immunotherapies. Herein, we review the current clinical advances and experimental findings supporting the utility of an anti-inflammatory approach to the treatment of solid malignancies. Gaining a better mechanistic understanding of the mode of action of anti-inflammatory agents and designing more effective treatment combinations would advance the clinical application of this therapeutic approach.
Figures
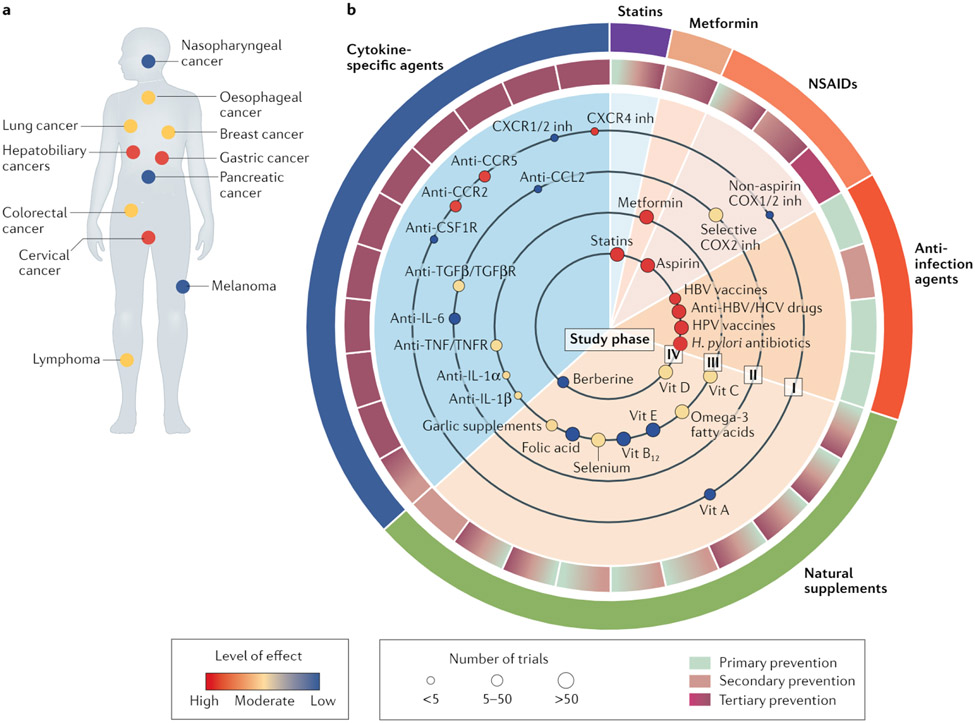
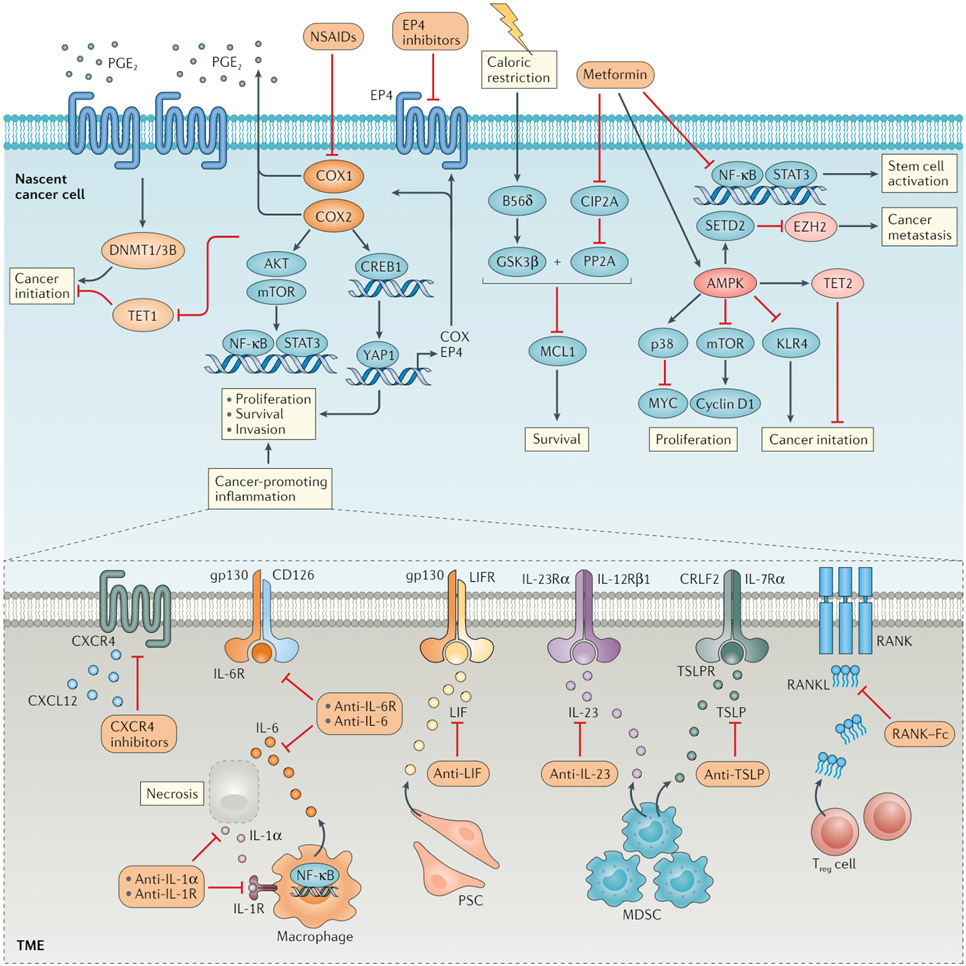
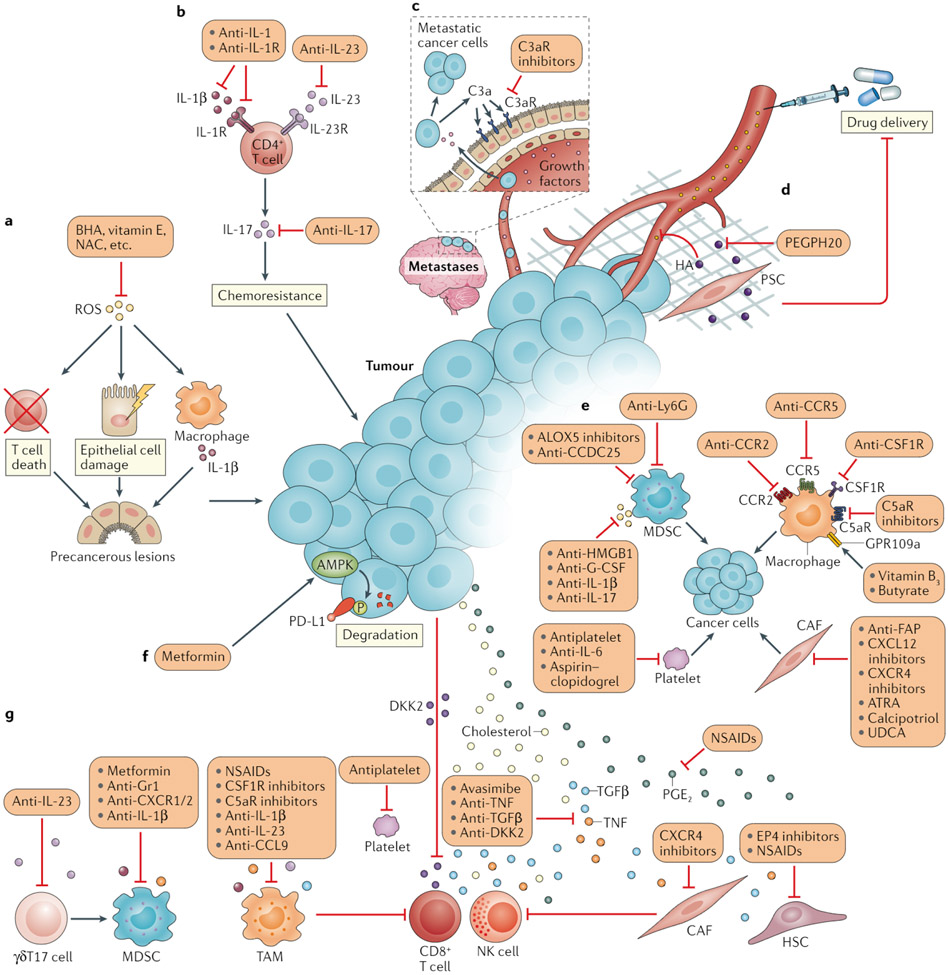
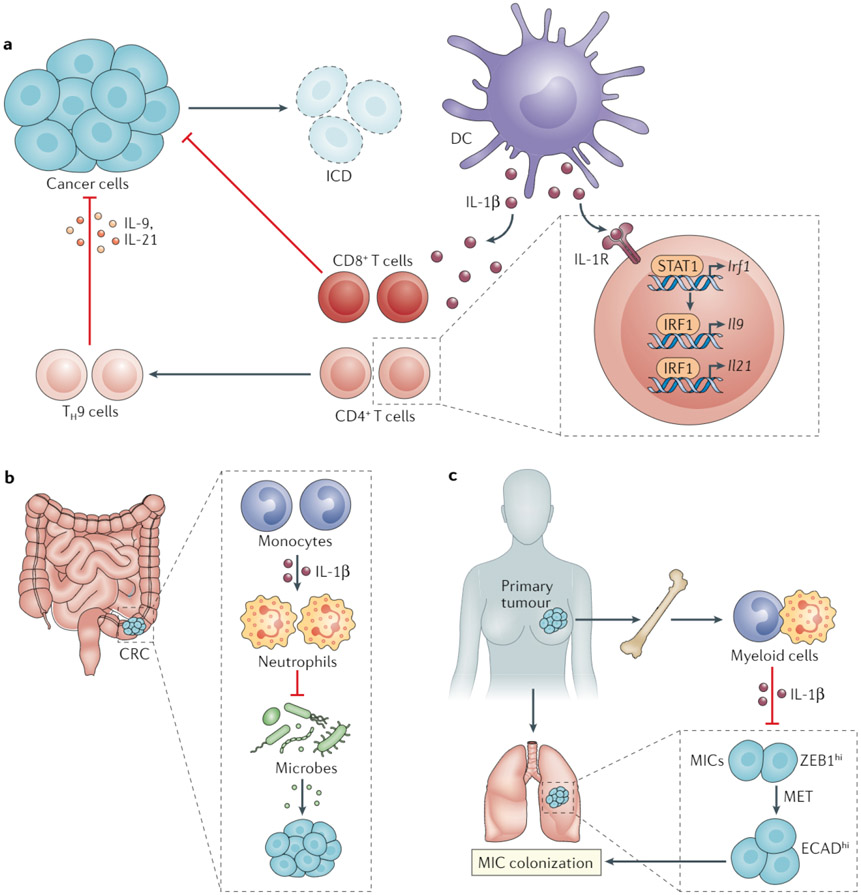
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
