

Sickle cell disease: progress towards combination drug therapy
- PMID: 33471938
- PMCID: PMC8282668
- DOI: 10.1111/bjh.17312
Sickle cell disease: progress towards combination drug therapy
Abstract
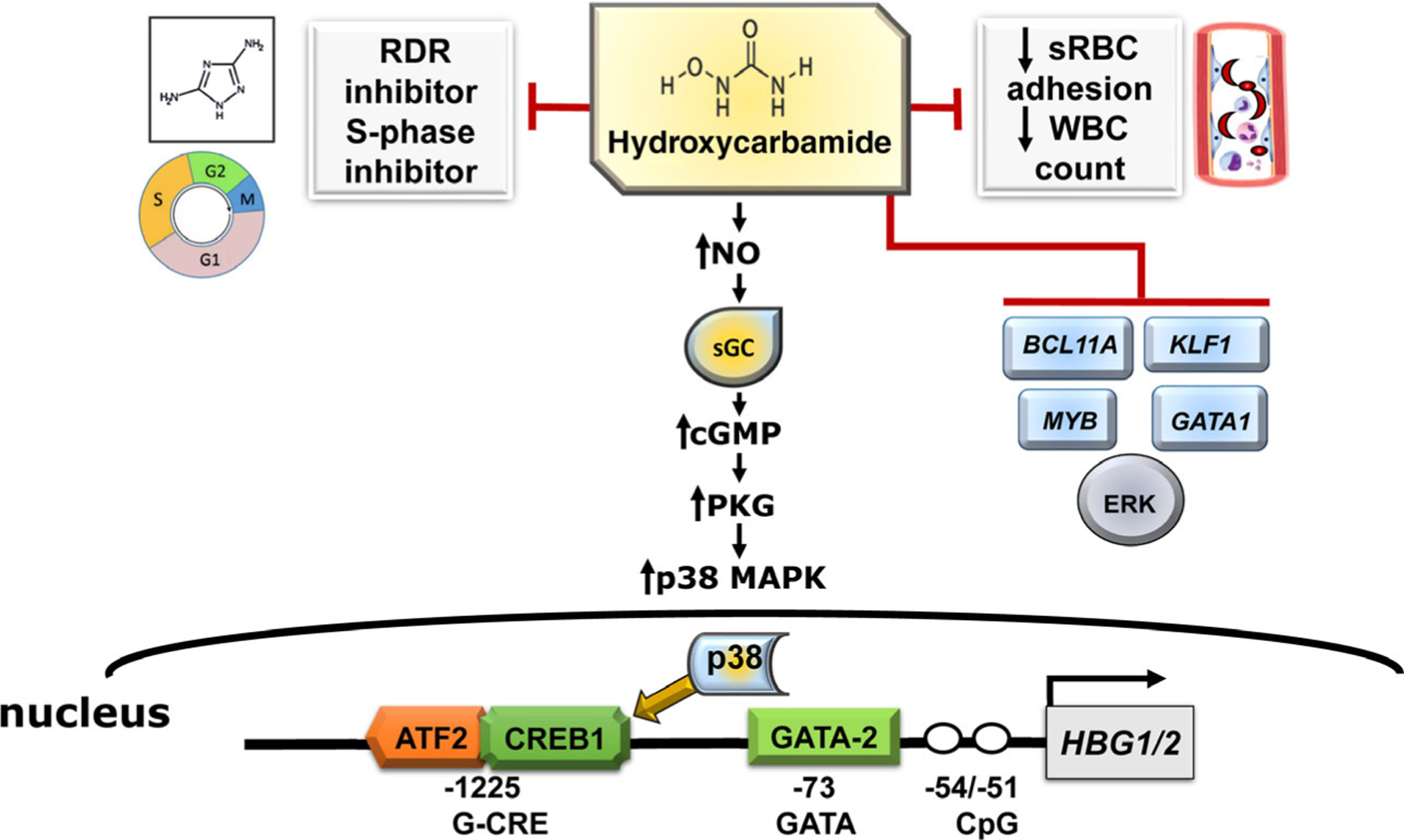
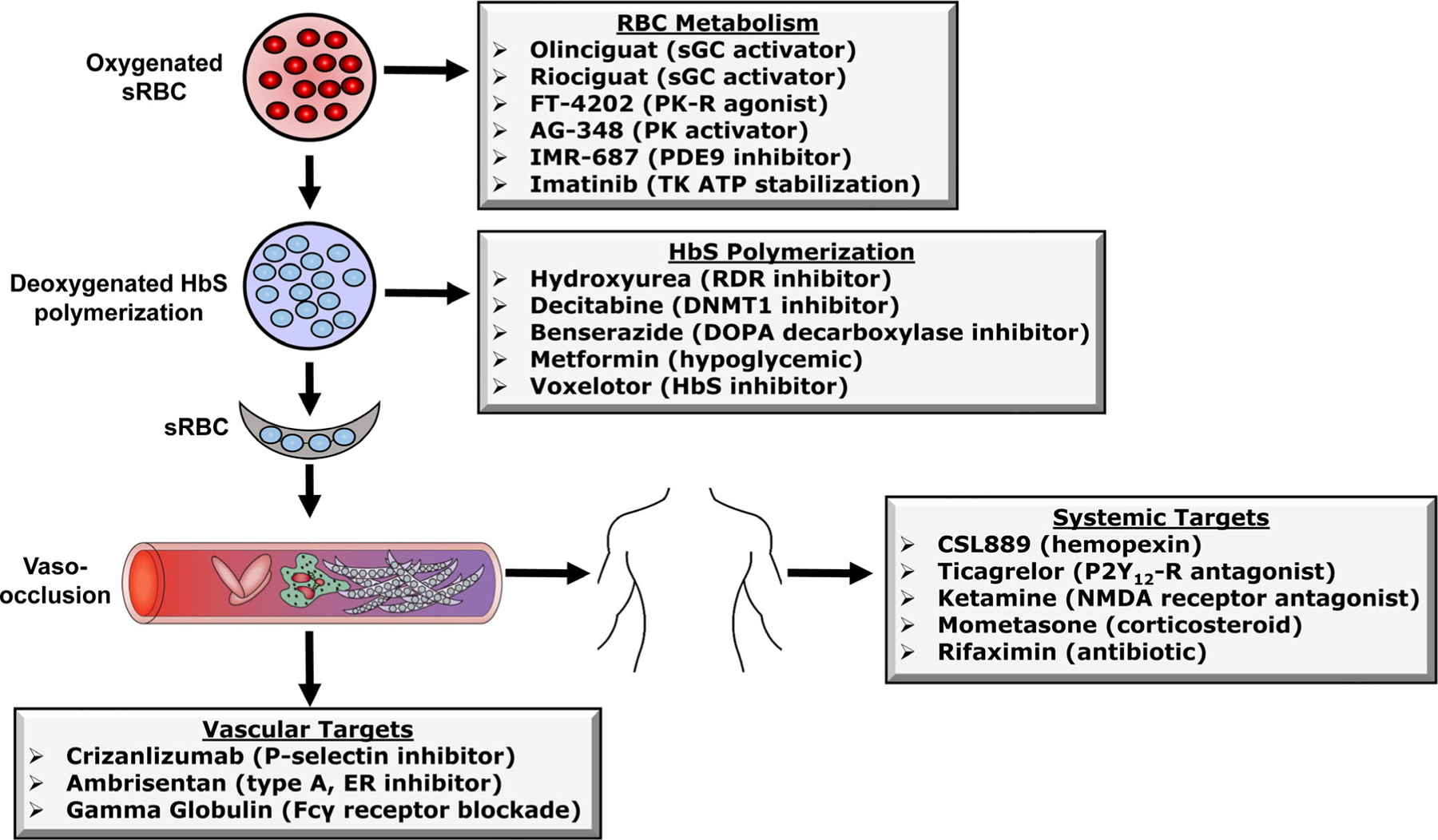
Dr. John Herrick described the first clinical case of sickle cell anaemia (SCA) in the United States in 1910. Subsequently, four decades later, Ingram and colleagues characterized the A to T substitution in DNA producing the GAG to GTG codon and replacement of glutamic acid with valine in the sixth position of the βS -globin chain. The establishment of Comprehensive Sickle Cell Centers in the United States in the 1970s was an important milestone in the development of treatment strategies and describing the natural history of sickle cell disease (SCD) comprised of genotypes including homozygous haemoglobin SS (HbSS), HbSβ0 thalassaemia, HbSC and HbSβ+ thalassaemia, among others. Early drug studies demonstrating effective treatments of HbSS and HbSβ0 thalassaemia, stimulated clinical trials to develop disease-specific therapies to induce fetal haemoglobin due to its ability to block HbS polymerization. Subsequently, hydroxycarbamide proved efficacious in adults with SCA and was Food and Drug Administration (FDA)-approved in 1998. After two decades of hydroxycarbamide use for SCD, there continues to be limited clinical acceptance of this chemotherapy drug, providing the impetus for investigators and pharmaceutical companies to develop non-chemotherapy agents. Investigative efforts to determine the role of events downstream of deoxy-HbS polymerization, such as endothelial cell activation, cellular adhesion, chronic inflammation, intravascular haemolysis and nitric oxide scavenging, have expanded drug targets which reverse the pathophysiology of SCD. After two decades of slow progress in the field, since 2018 three new drugs were FDA-approved for SCA, but research efforts to develop treatments continue. Currently over 30 treatment intervention trials are in progress to investigate a wide range of agents acting by complementary mechanisms, providing the rationale for ushering in the age of effective and safe combination drug therapy for SCD. Parallel efforts to develop curative therapies using haematopoietic stem cell transplant and gene therapy provide individuals with SCD multiple treatment options. We will discuss progress made towards drug development and potential combination drug therapy for SCD with the standard of care hydroxycarbamide.
Keywords: combination drug therapy; fetal haemoglobin; gene therapy; haematopoietic stem cell transplant; sickle cell anaemia; sickle cell disease.
© 2021 British Society for Haematology and John Wiley & Sons Ltd.
Figures
References
-
- Kauf TL, Coates TD, Huazhi L, Mody-Patel N, Hartzema AG. The cost of health care for children and adults with sickle cell disease. Am J Hematol. 2009;84:323–7. - PubMed
-
- Ashley-Koch A, Yang Q, Olney RS. Sickle hemoglobin (HbS) allele and sickle cell disease: a HuGE review. Am J Epidemiol. 2000;151:839–45. - PubMed
-
- Gardner K, Douiri A, Drasar E, Allman M, Mwirigi A, Awogbade M, et al. Survival in adults with sickle cell disease in a high-income setting. Blood. 2016;128:1436–8. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
