

A platform for experimental precision medicine: The extended BXD mouse family
- PMID: 33472028
- PMCID: PMC7979527
- DOI: 10.1016/j.cels.2020.12.002
A platform for experimental precision medicine: The extended BXD mouse family
Abstract
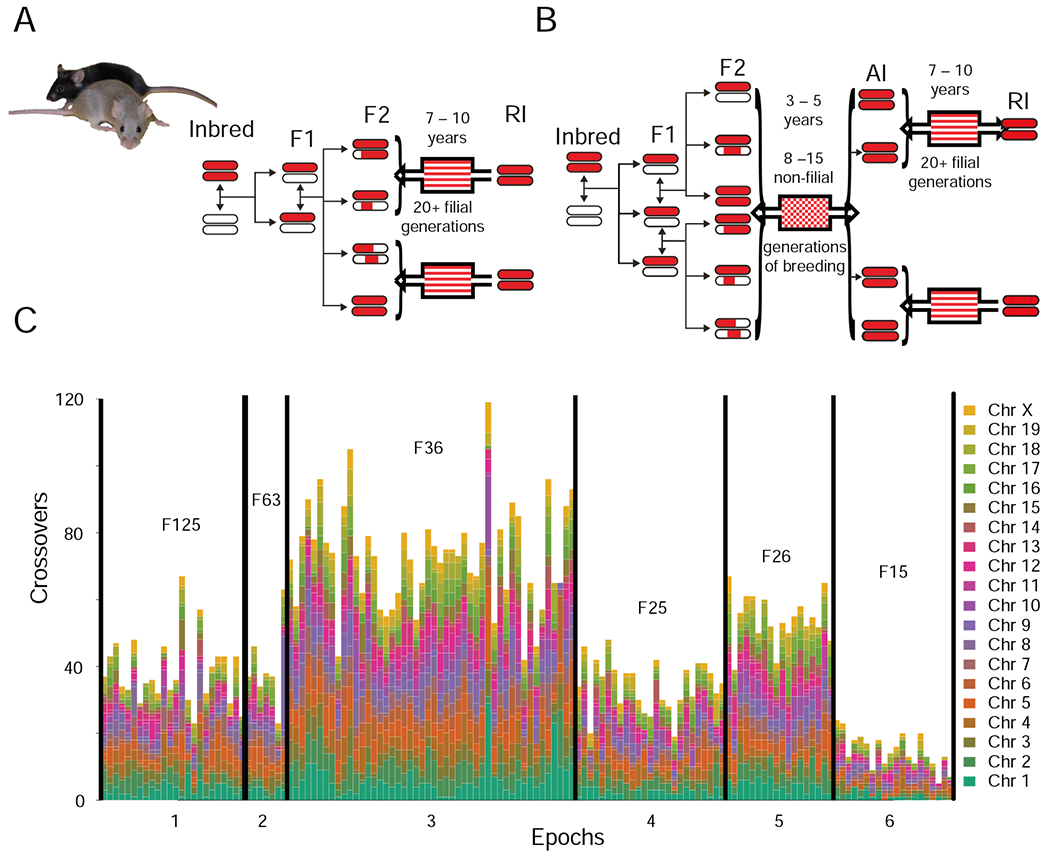
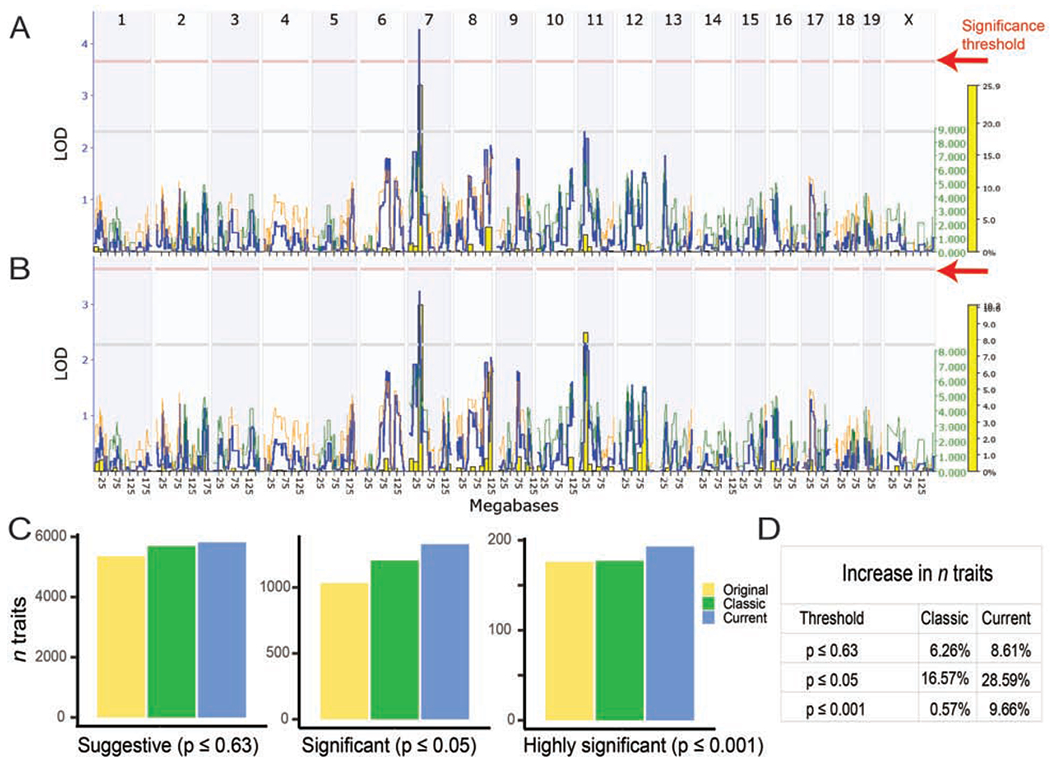
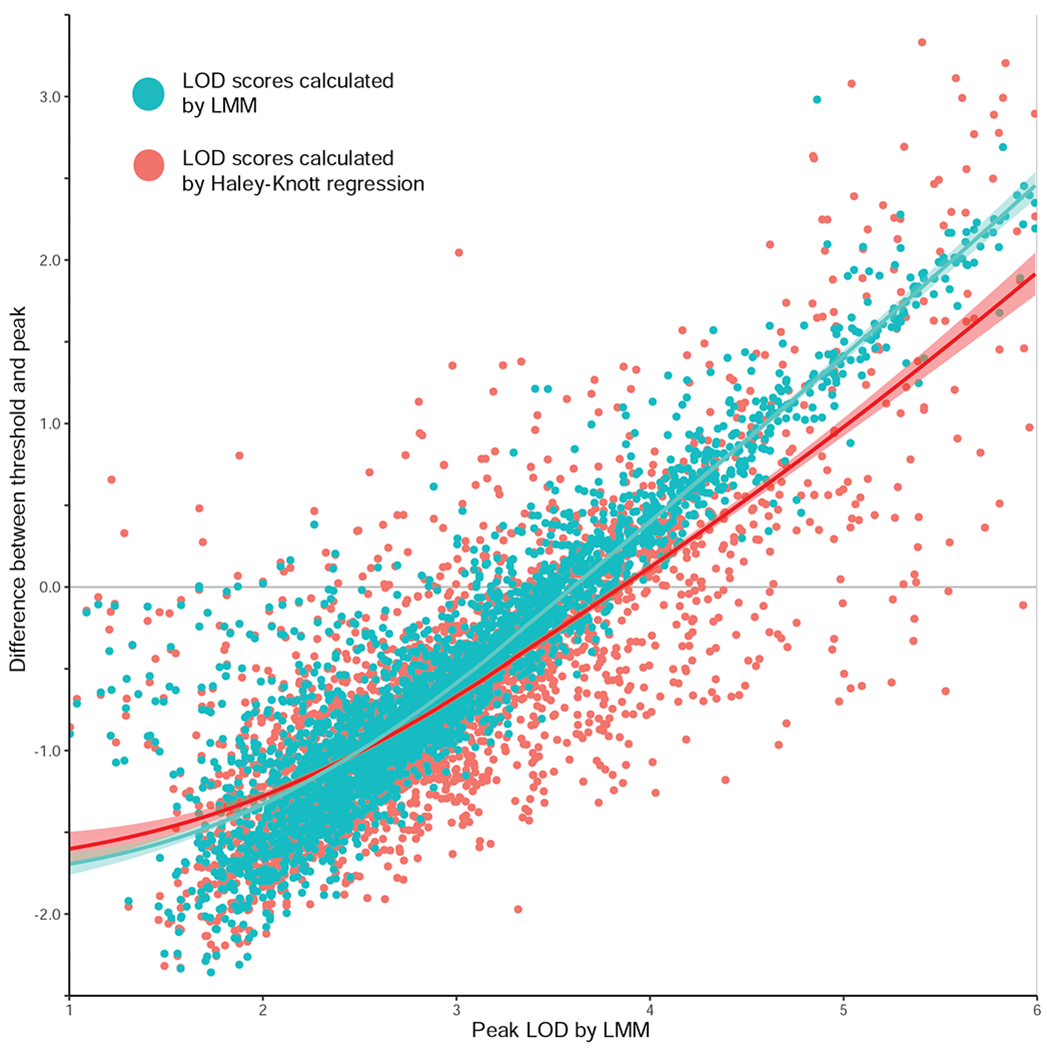
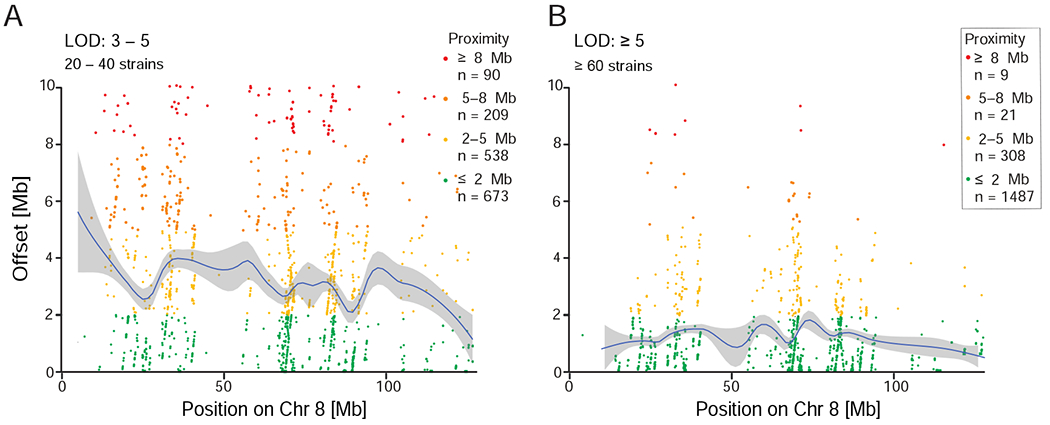
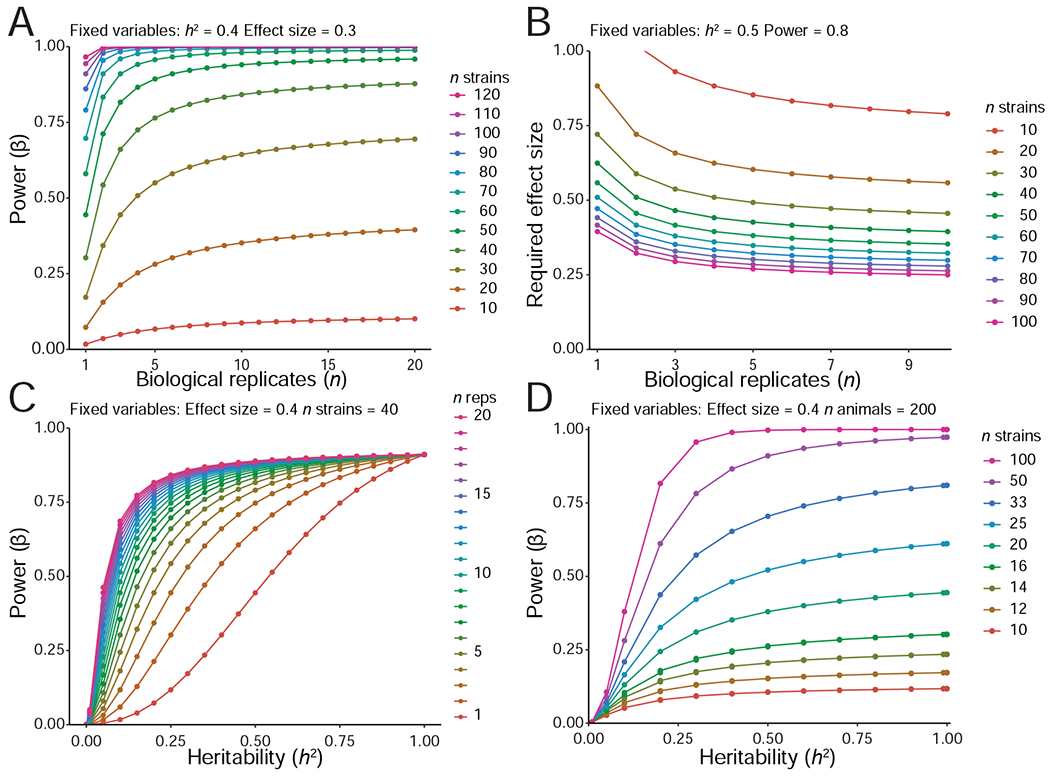
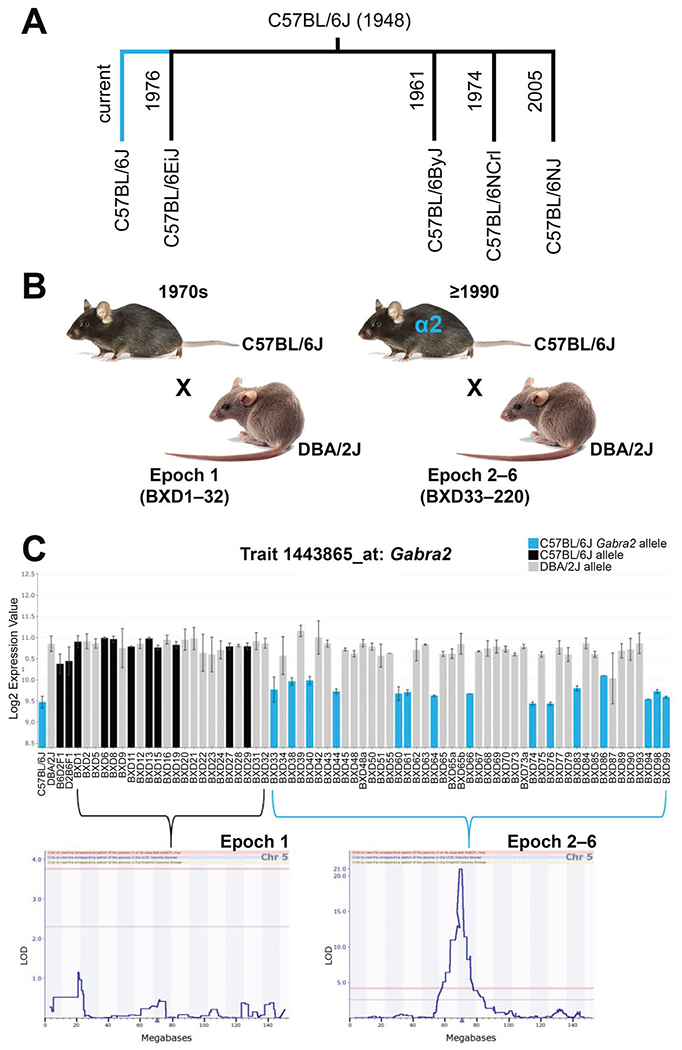
The challenge of precision medicine is to model complex interactions among DNA variants, phenotypes, development, environments, and treatments. We address this challenge by expanding the BXD family of mice to 140 fully isogenic strains, creating a uniquely powerful model for precision medicine. This family segregates for 6 million common DNA variants-a level that exceeds many human populations. Because each member can be replicated, heritable traits can be mapped with high power and precision. Current BXD phenomes are unsurpassed in coverage and include much omics data and thousands of quantitative traits. BXDs can be extended by a single-generation cross to as many as 19,460 isogenic F1 progeny, and this extended BXD family is an effective platform for testing causal modeling and for predictive validation. BXDs are a unique core resource for the field of experimental precision medicine.
Keywords: GXE; complex trait; gene mapping; personalized medicine; power calculation; recombinant inbred strains; systems biology; systems genetics.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
References
-
- Ashbrook DG, Delprato A, Grellmann C, Klein M, Wetzel R, Overall RW, and Badea A (2014b). Transcript co-variance with Nestin in two mouse genetic reference populations identifies Lef1 as a novel candidate regulator of neural precursor cell proliferation in the adult hippocampus. Front Neurosci 8, 418. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
