

Transcriptome alterations in myotonic dystrophy frontal cortex
- PMID: 33472074
- PMCID: PMC9272850
- DOI: 10.1016/j.celrep.2020.108634
Transcriptome alterations in myotonic dystrophy frontal cortex
Abstract
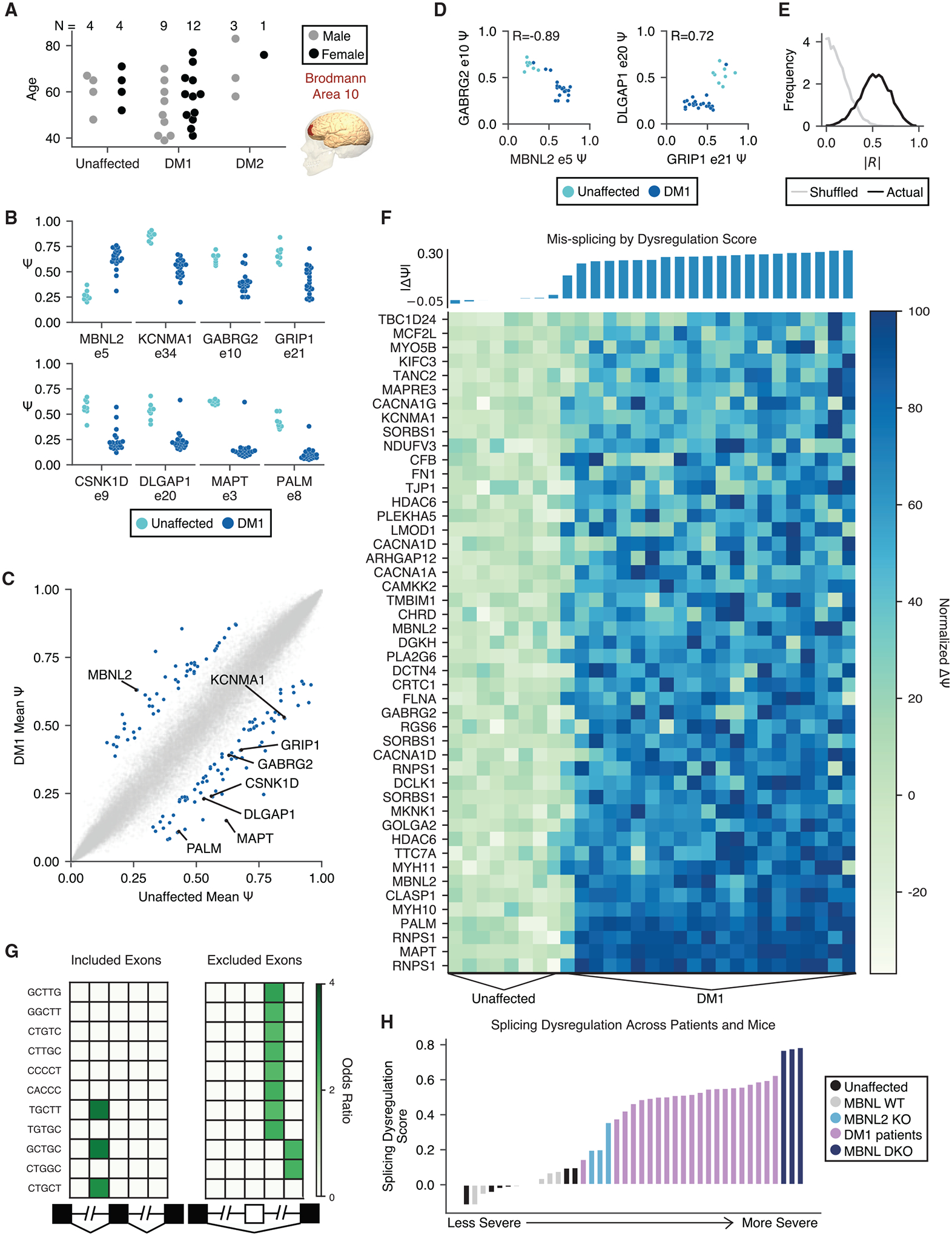
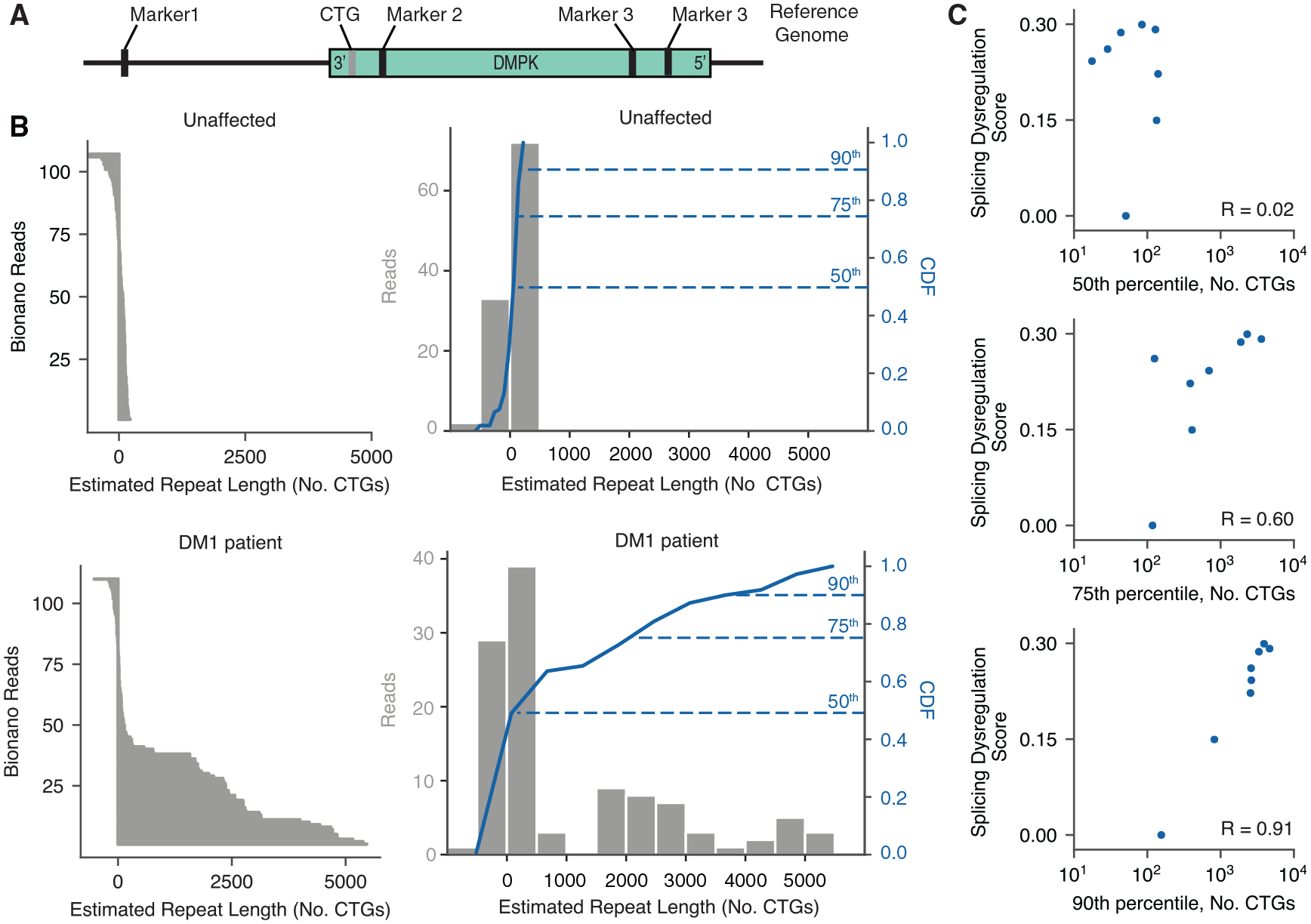
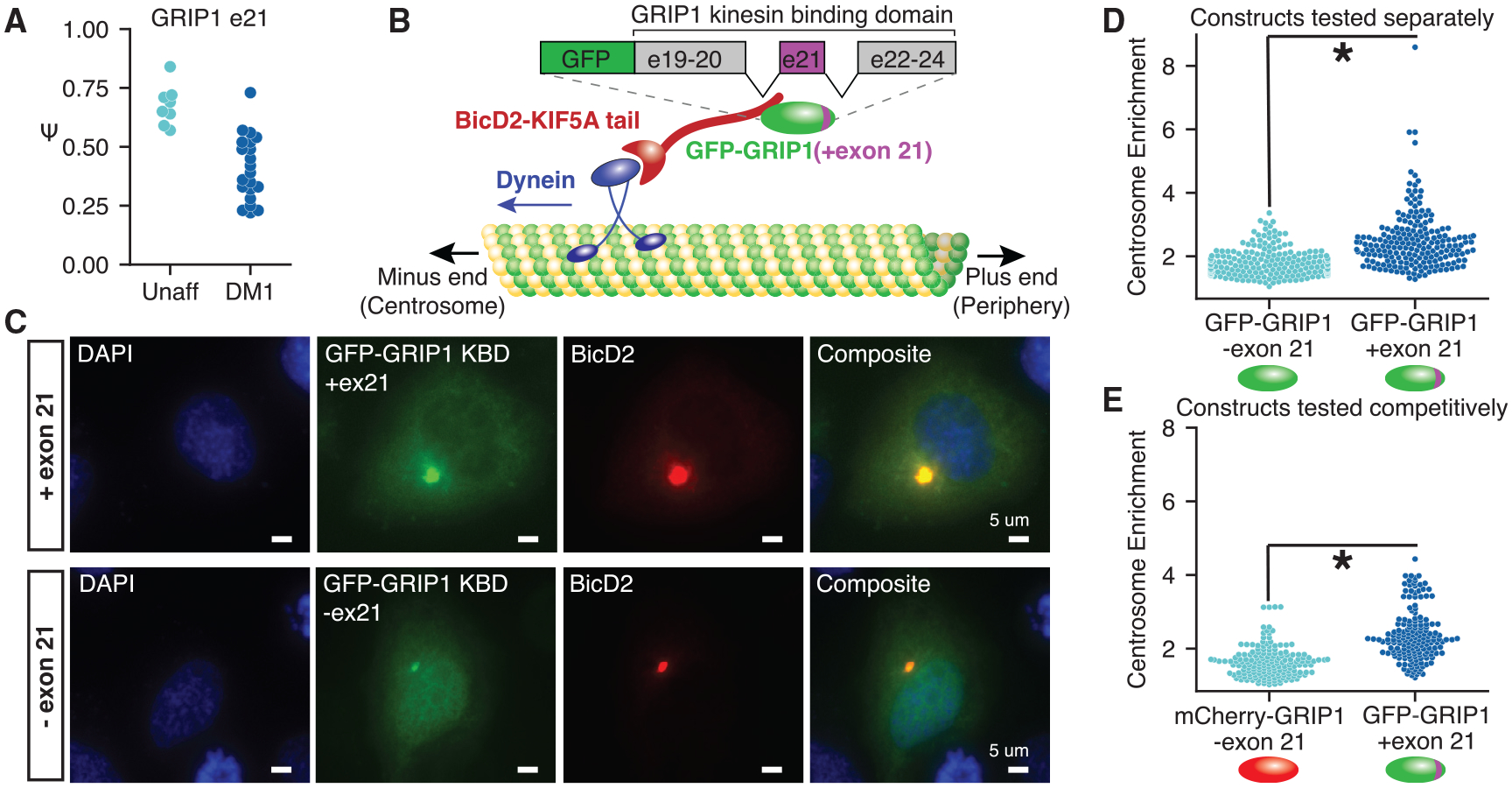
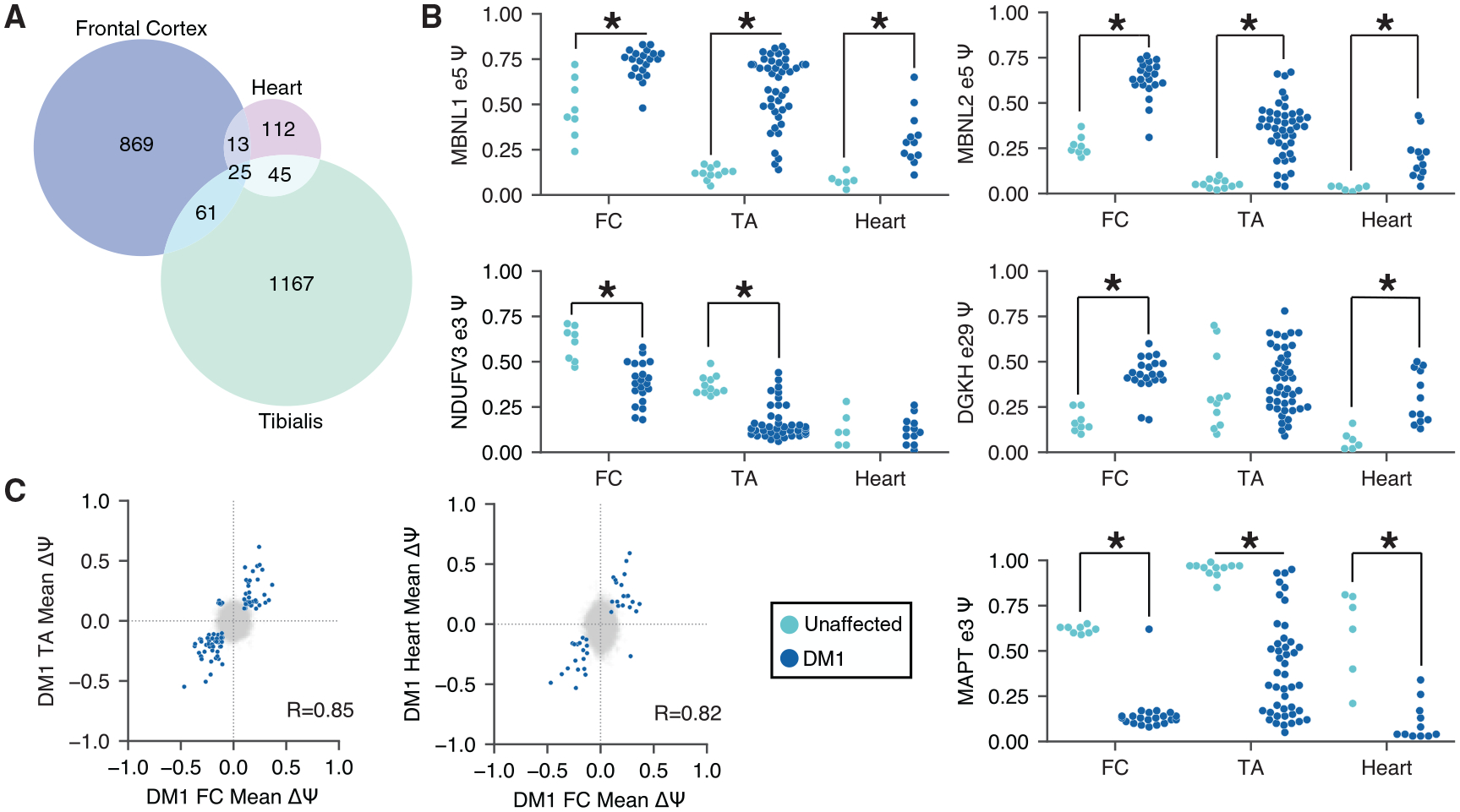
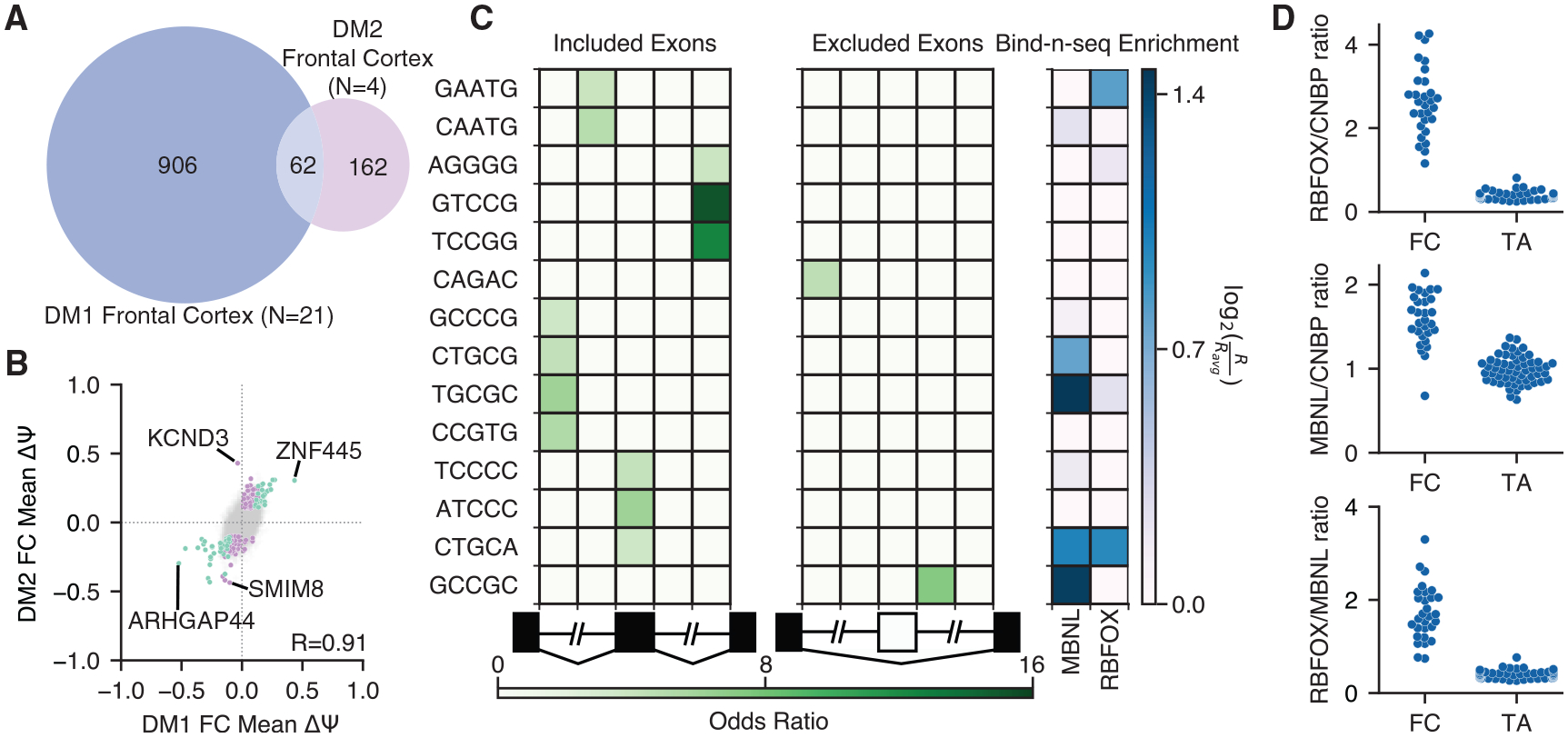
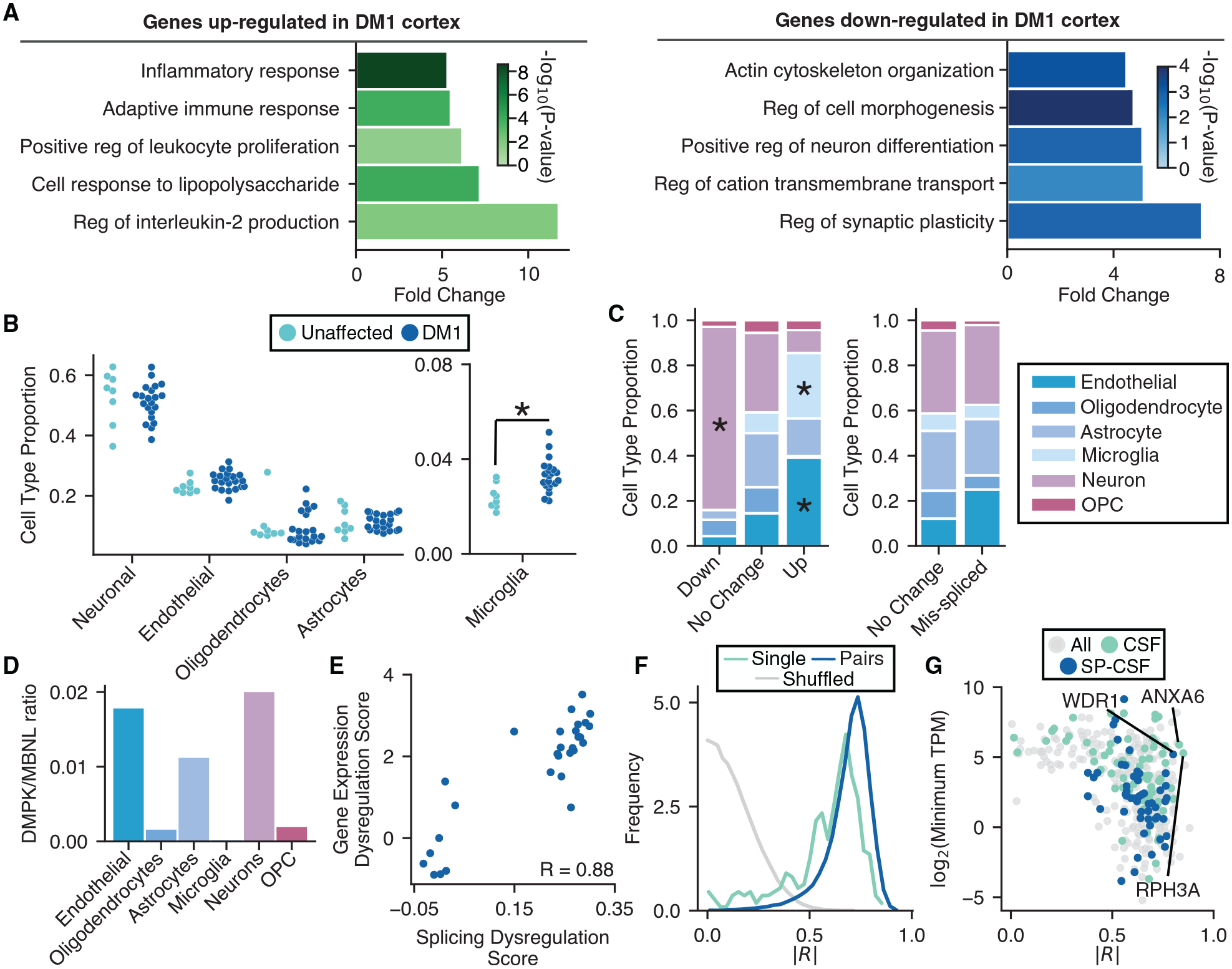
Myotonic dystrophy (DM) is caused by expanded CTG/CCTG repeats, causing symptoms in skeletal muscle, heart, and central nervous system (CNS). CNS issues are debilitating and include hypersomnolence, executive dysfunction, white matter atrophy, and neurofibrillary tangles. Here, we generate RNA-seq transcriptomes from DM and unaffected frontal cortex and identify 130 high-confidence splicing changes, most occurring only in cortex, not skeletal muscle or heart. Mis-spliced exons occur in neurotransmitter receptors, ion channels, and synaptic scaffolds, and GRIP1 mis-splicing modulates kinesin association. Optical mapping of expanded CTG repeats reveals extreme mosaicism, with some alleles showing >1,000 CTGs. Mis-splicing severity correlates with CTG repeat length across individuals. Upregulated genes tend to be microglial and endothelial, suggesting neuroinflammation, and downregulated genes tend to be neuronal. Many gene expression changes strongly correlate with mis-splicing, suggesting candidate biomarkers of disease. These findings provide a framework for mechanistic and therapeutic studies of the DM CNS.
Copyright © 2020 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
References
-
- Andrews S (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data. Babraham Bioinformatics http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
