

VELCRO-IP RNA-seq reveals ribosome expansion segment function in translation genome-wide
- PMID: 33472078
- PMCID: PMC8270675
- DOI: 10.1016/j.celrep.2020.108629
VELCRO-IP RNA-seq reveals ribosome expansion segment function in translation genome-wide
Abstract
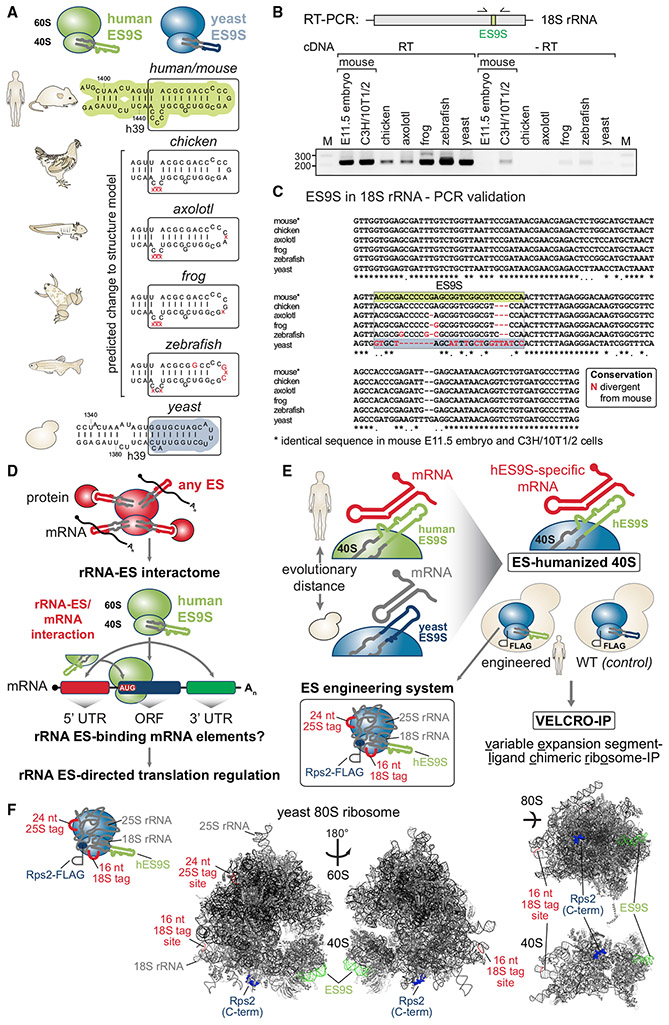
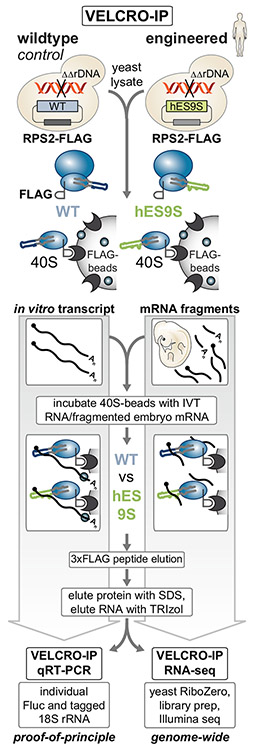
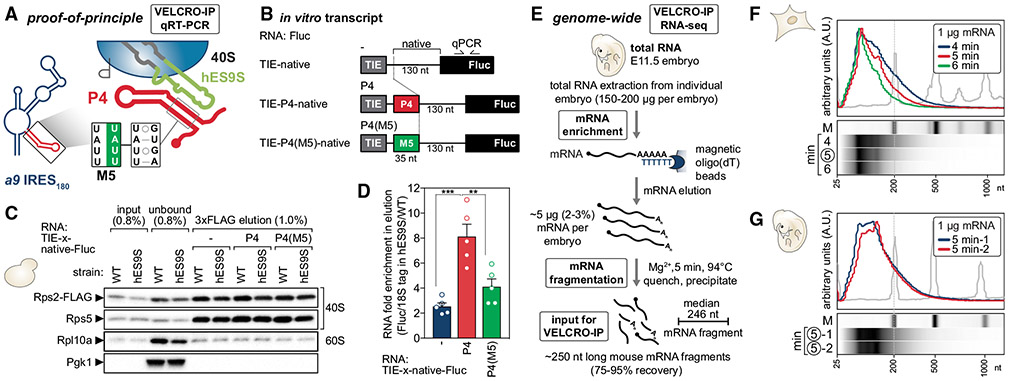
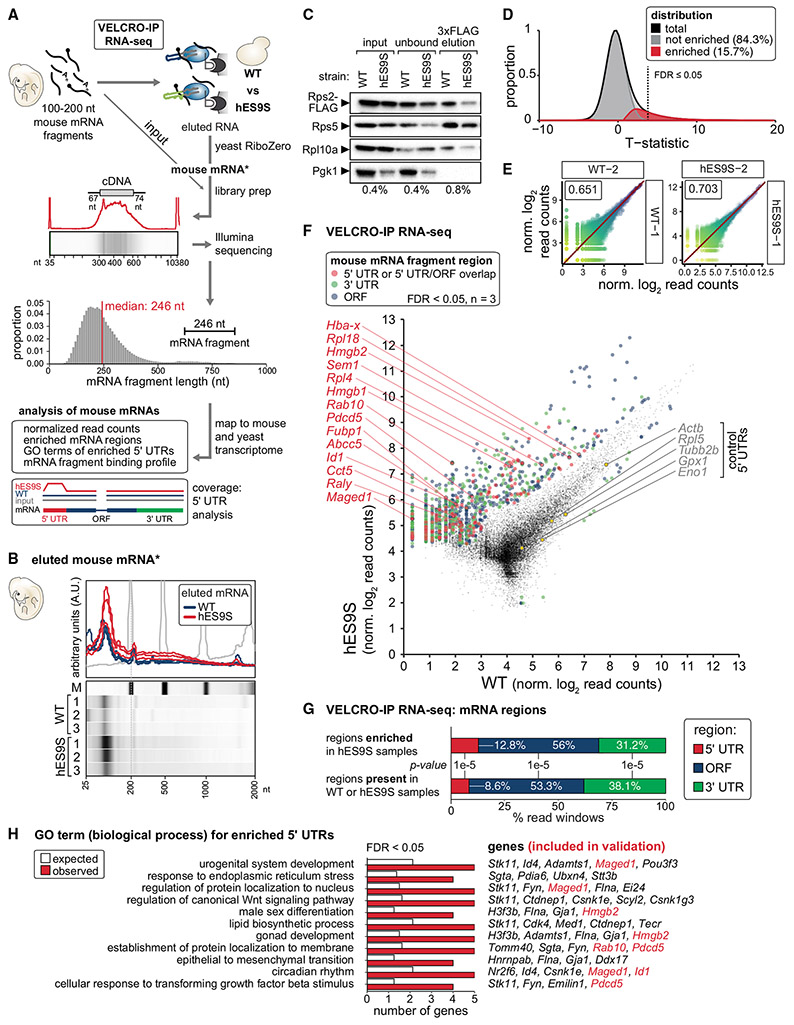
Roles for ribosomal RNA (rRNA) in gene regulation remain largely unexplored. With hundreds of rDNA units positioned across multiple loci, it is not possible to genetically modify rRNA in mammalian cells, hindering understanding of ribosome function. It remains elusive whether expansion segments (ESs), tentacle-like rRNA extensions that vary in sequence and size across eukaryotic evolution, may have functional roles in translation control. Here, we develop variable expansion segment-ligand chimeric ribosome immunoprecipitation RNA sequencing (VELCRO-IP RNA-seq), a versatile methodology to generate species-adapted ESs and to map specific mRNA regions across the transcriptome that preferentially associate with ESs. Application of VELCRO-IP RNA-seq to a mammalian ES, ES9S, identified a large array of transcripts that are selectively recruited to ribosomes via an ES. We further characterize a set of 5' UTRs that facilitate cap-independent translation through ES9S-mediated ribosome binding. Thus, we present a technology for studying the enigmatic ESs of the ribosome, revealing their function in gene-specific translation.
Keywords: ES9S; RNA fragment mapping; RNA sequencing; RNA stem-loop structure; internal initiation; mRNA translation; mouse embryo; rRNA expansion segment; ribosome engineering; yeast.
Copyright © 2020 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests K.L., G.W.B., and M.B. are inventors on patents and submitted provisional patent applications related to the Hoxa9 P4 stem-loop and RNA therapeutics and to their various uses.
Figures
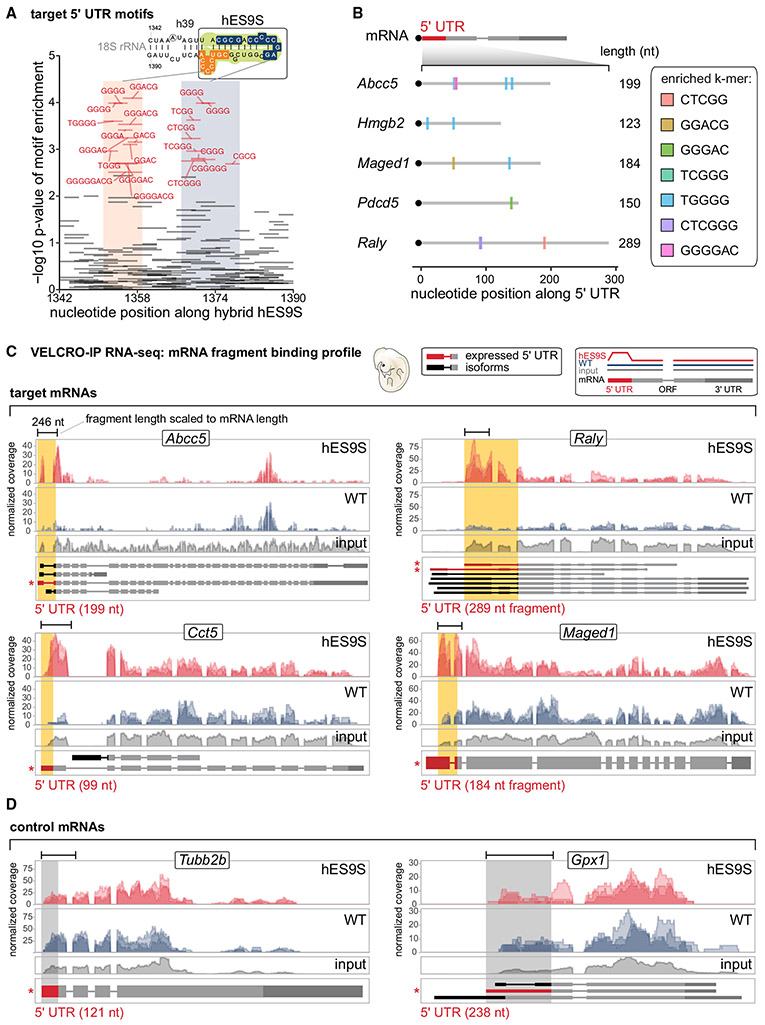
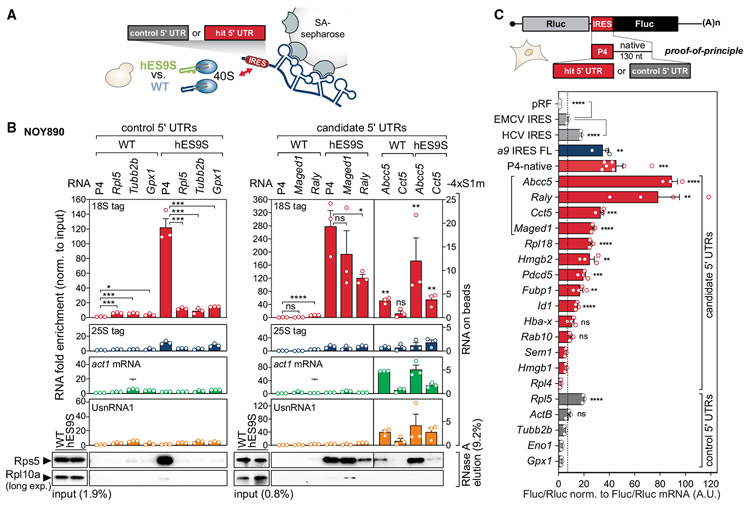
References
-
- Alexa A, Rahnenfiihrer J, and Lengauer T (2006). Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 22, 1600–1607. - PubMed
-
- Anger AM, Armache JP, Berninghausen O, Habeck M, Subklewe M, Wilson DN, and Beckmann R (2013). Structures of the human and Drosophila 80S ribosome. Nature 497, 80–85. - PubMed
-
- Appels R, Gerlach WL, Dennis ES, Swift H, and Peacock WJ (1980). Molecular and chromosomal organization of DNA sequences coding for the ribosomal RNAs in cereals. Chromosoma 78, 293–311.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
