

The RNA-binding protein SFPQ preserves long-intron splicing and regulates circRNA biogenesis in mammals
- PMID: 33476259
- PMCID: PMC7819710
- DOI: 10.7554/eLife.63088
The RNA-binding protein SFPQ preserves long-intron splicing and regulates circRNA biogenesis in mammals
Abstract
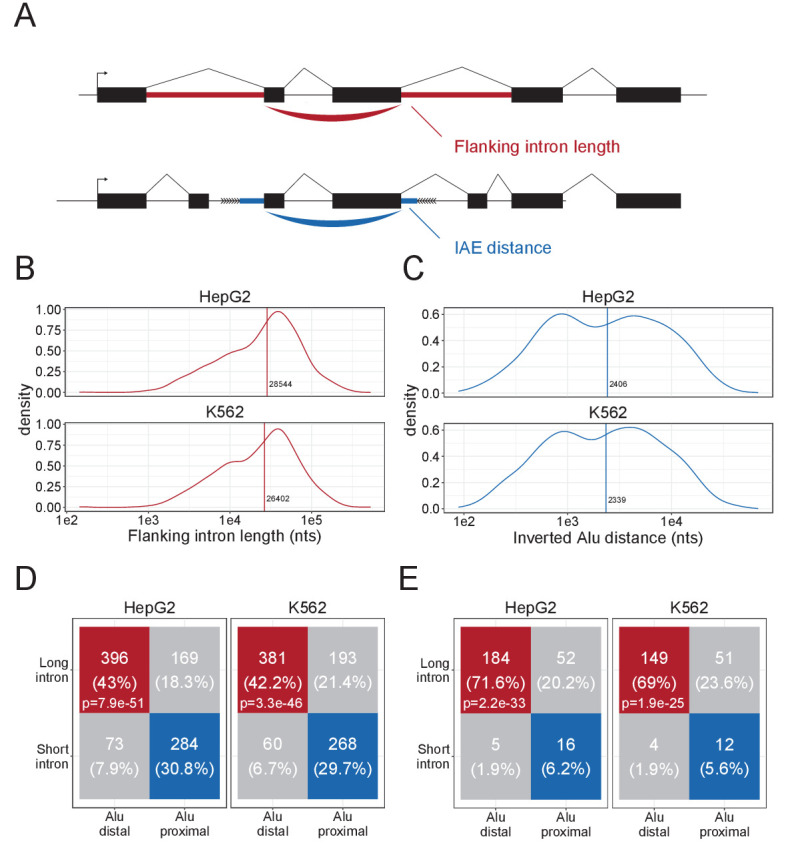
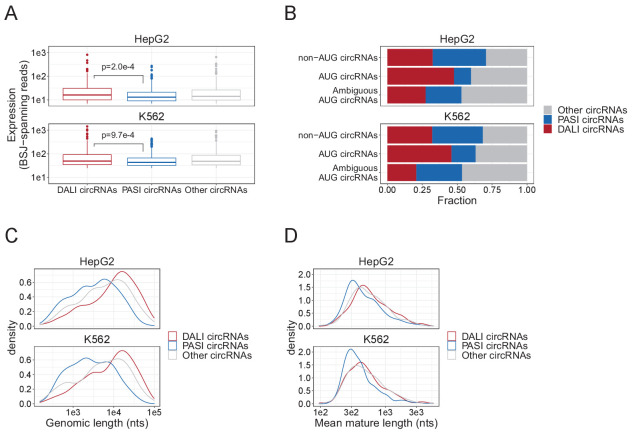
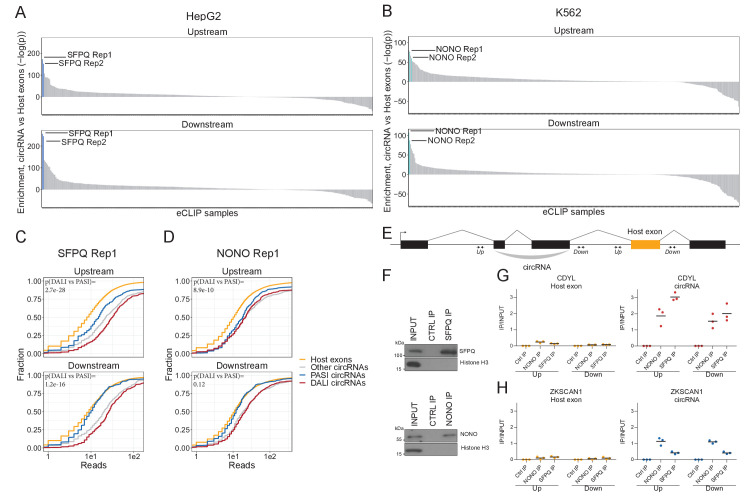
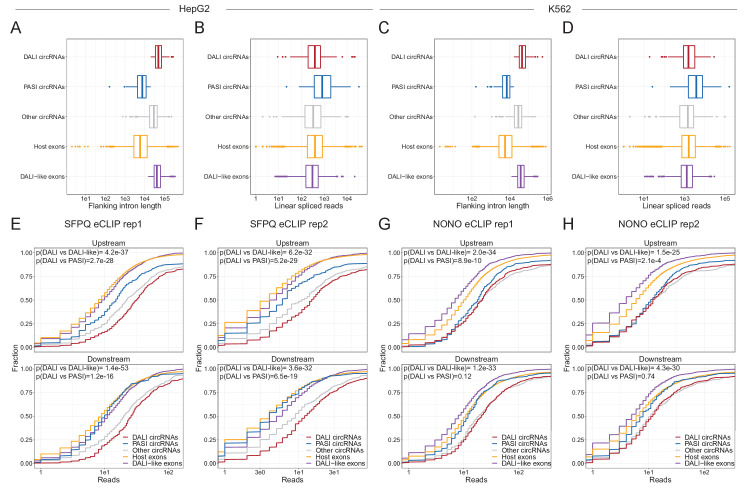
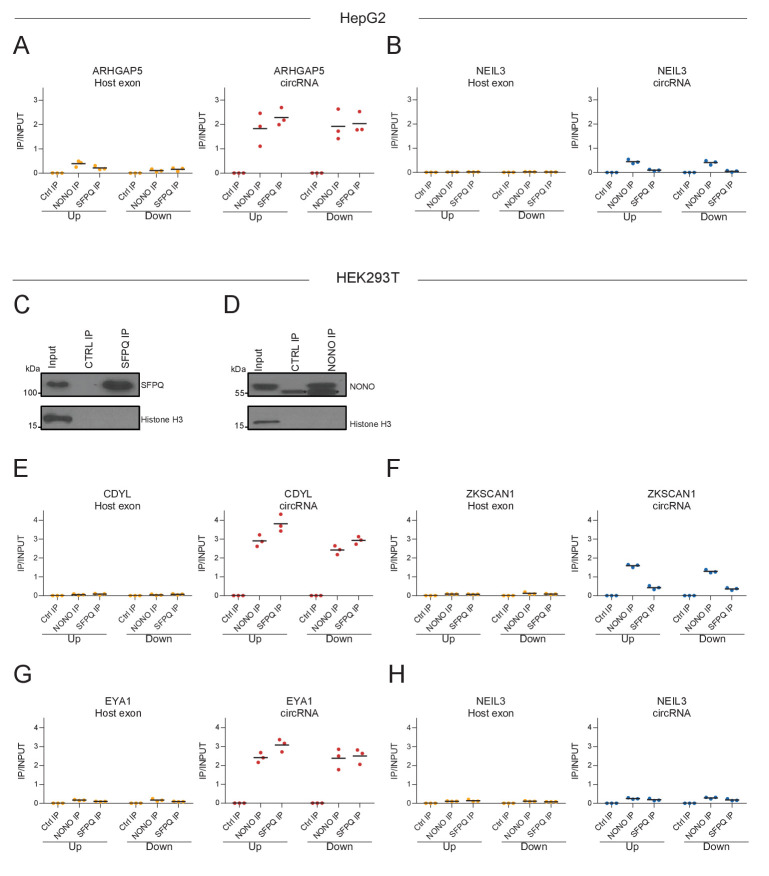
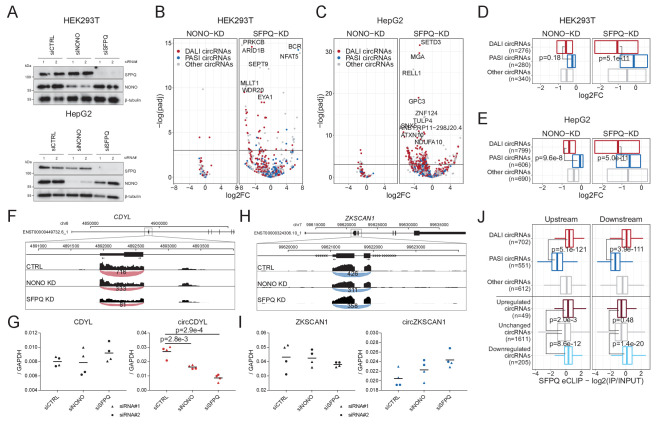
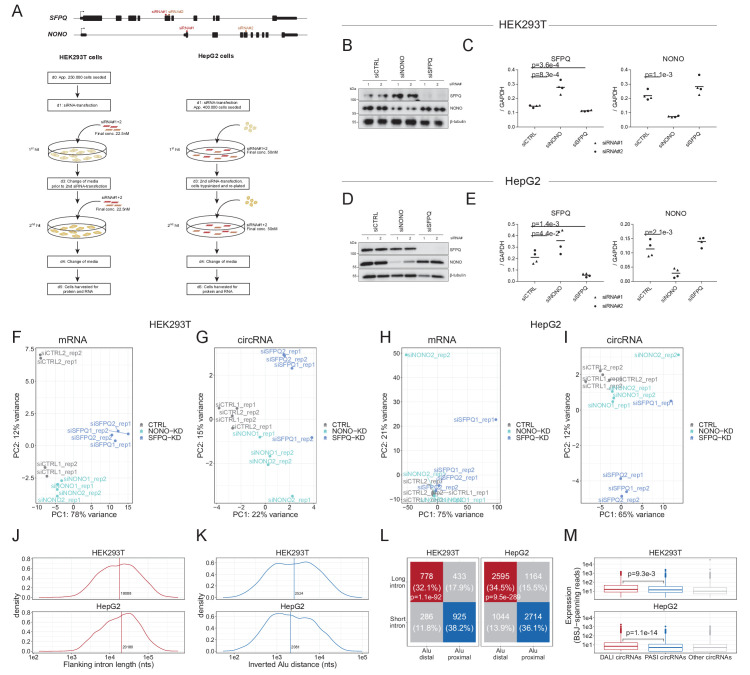
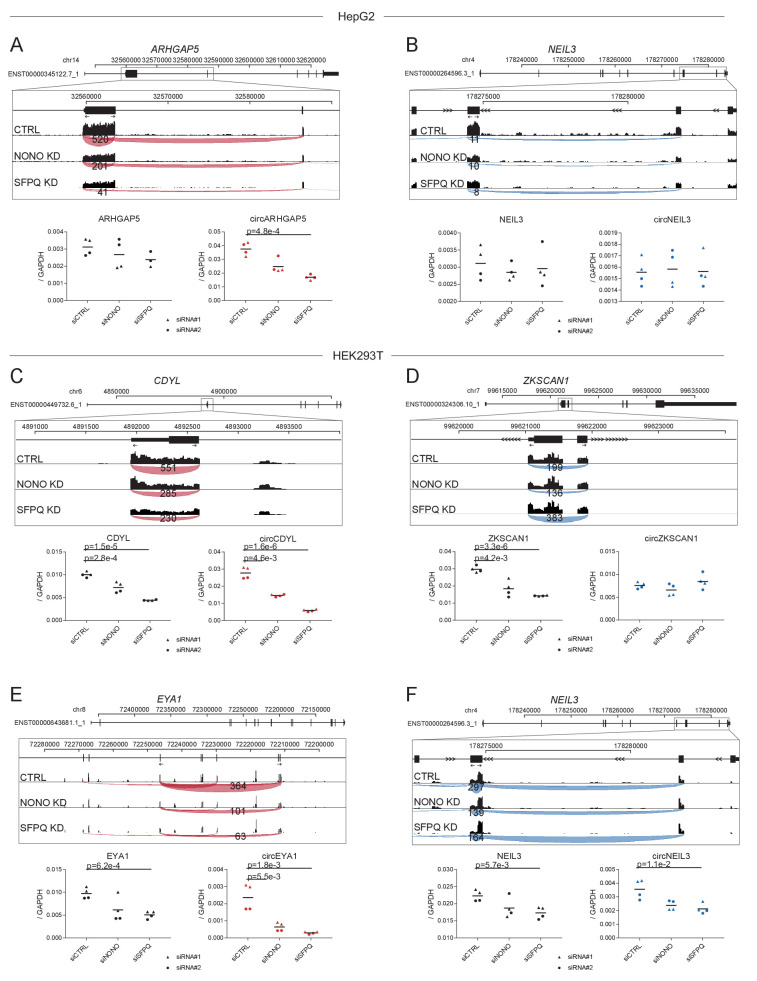
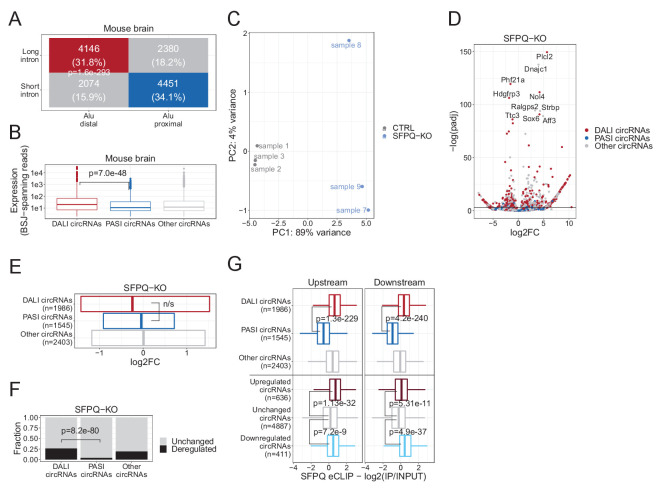
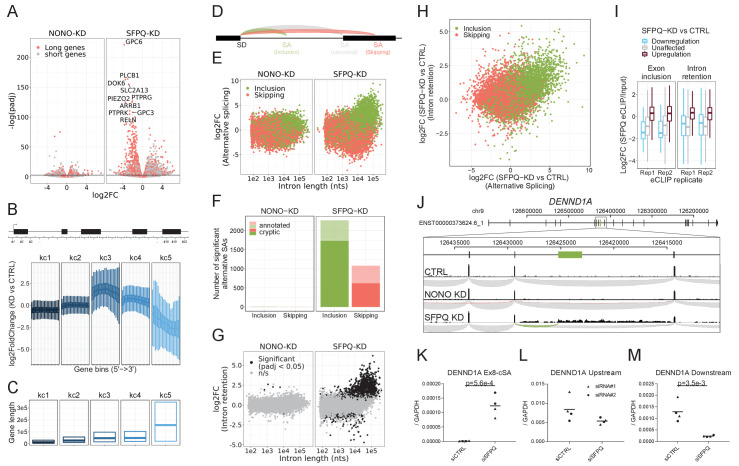
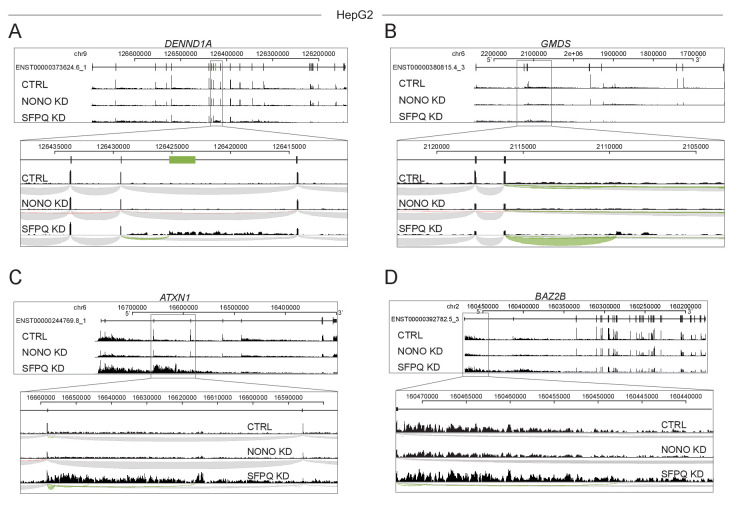
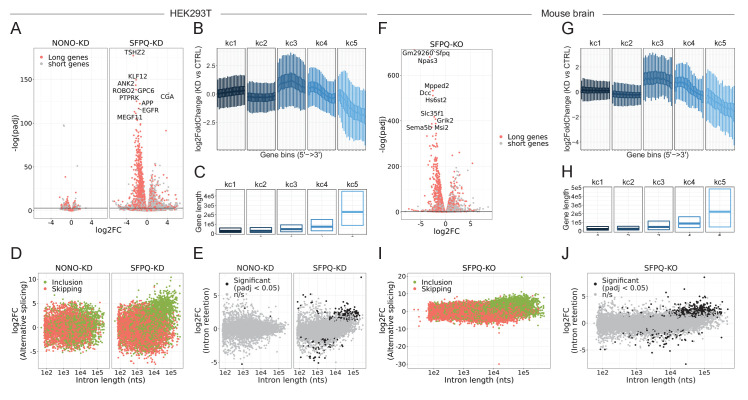
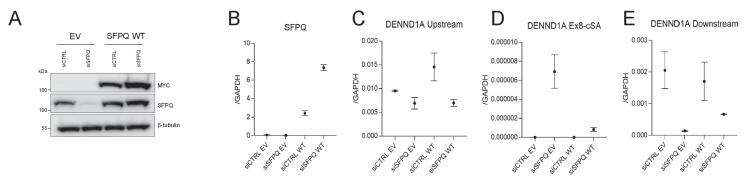
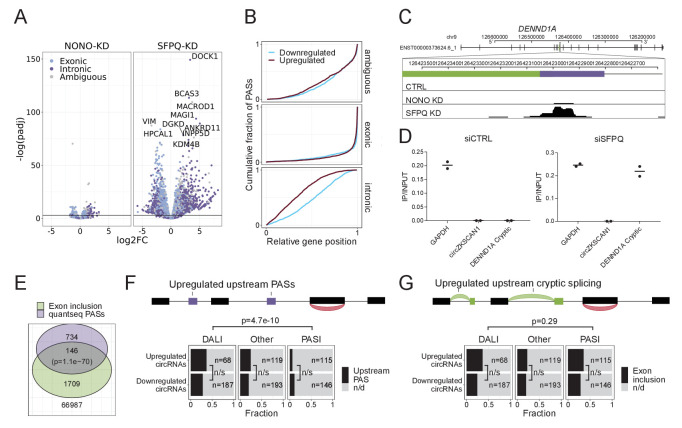
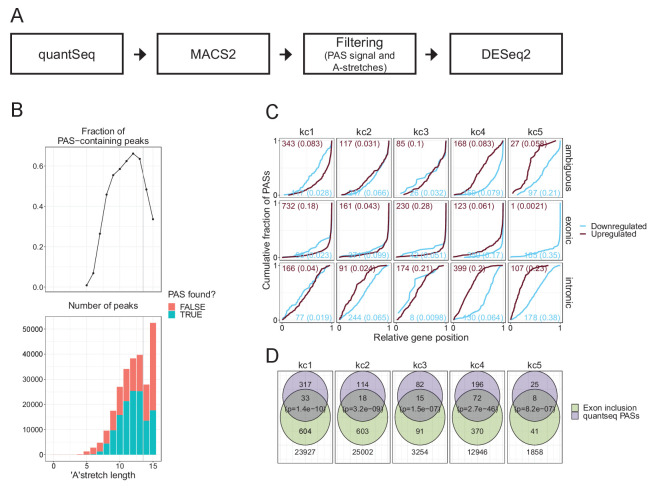
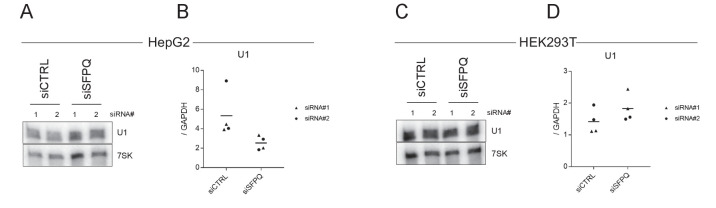
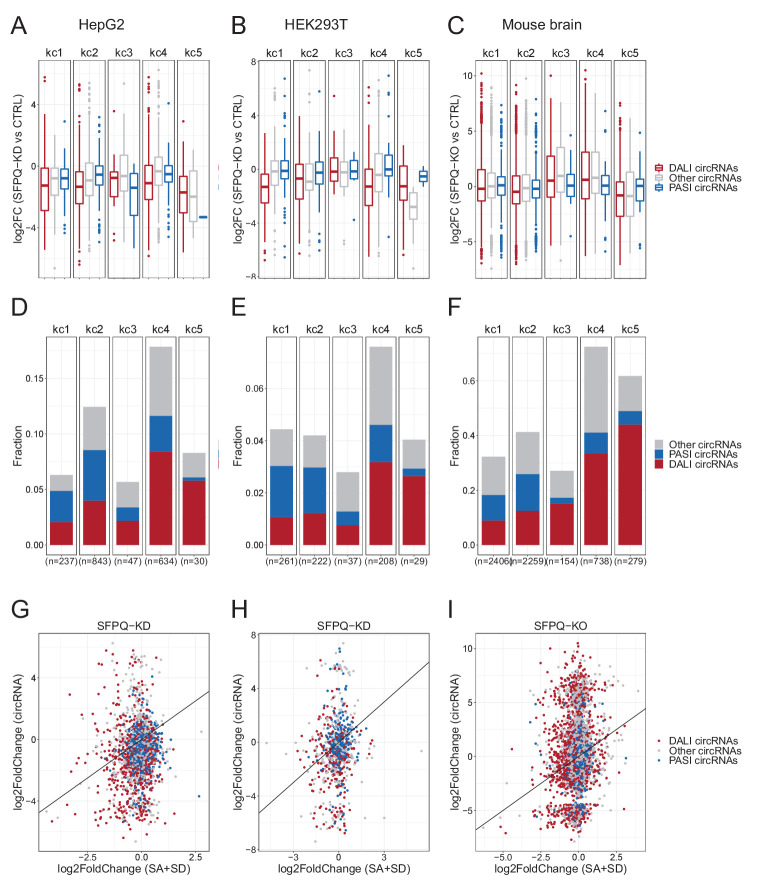
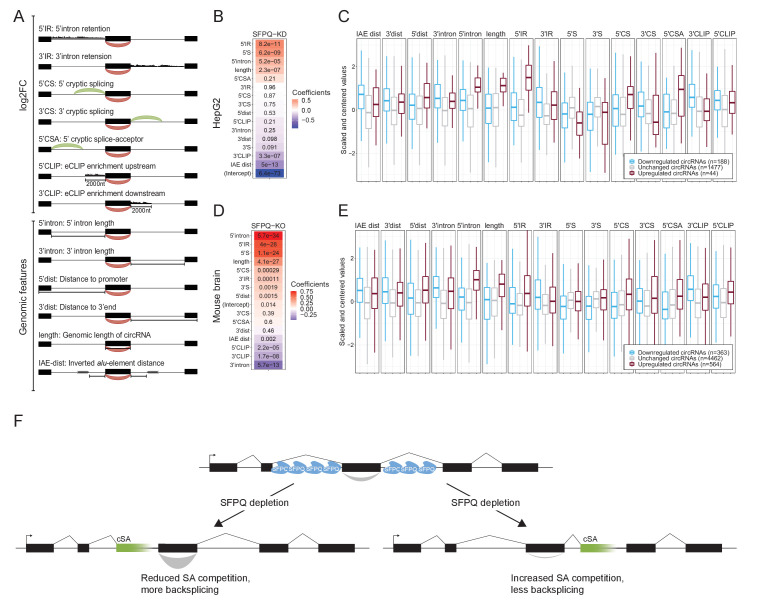
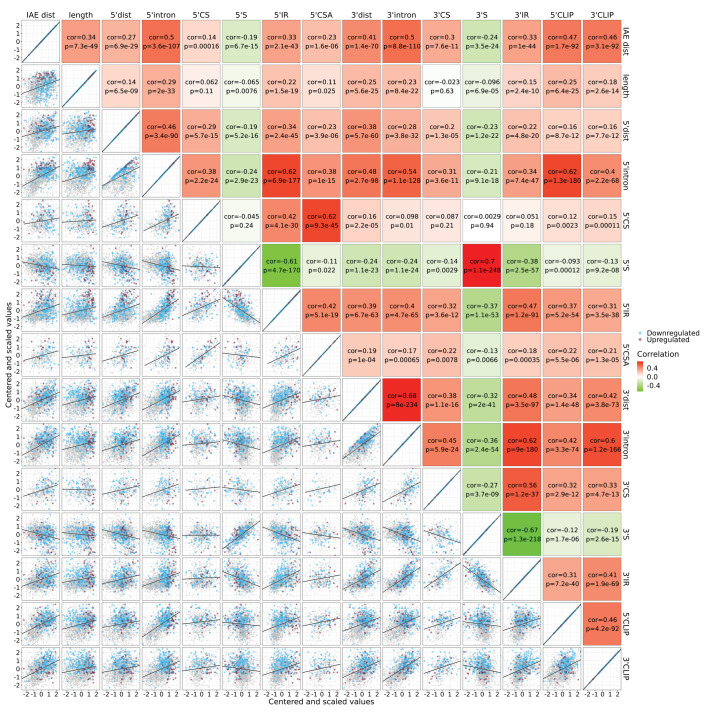
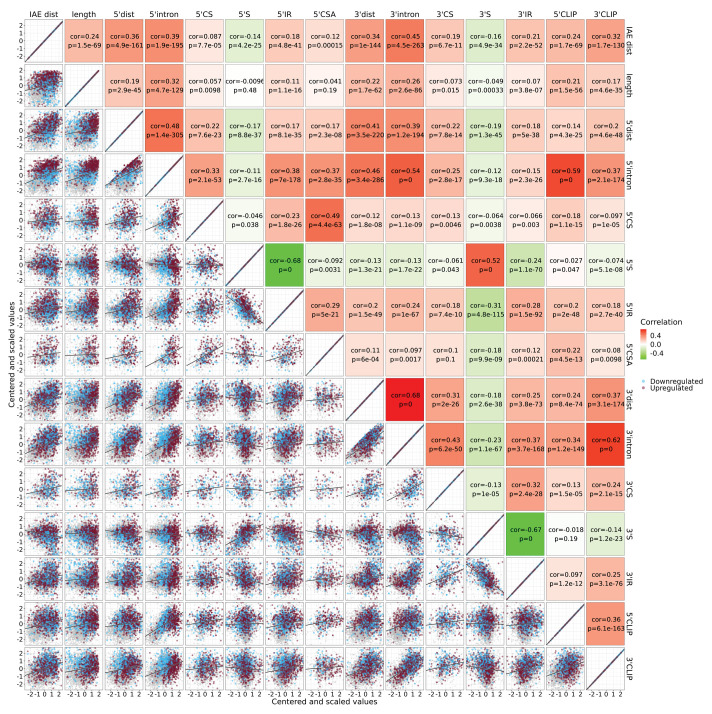
Circular RNAs (circRNAs) represent an abundant and conserved entity of non-coding RNAs; however, the principles of biogenesis are currently not fully understood. Here, we identify two factors, splicing factor proline/glutamine rich (SFPQ) and non-POU domain-containing octamer-binding protein (NONO), to be enriched around circRNA loci. We observe a subclass of circRNAs, coined DALI circRNAs, with distal inverted Alu elements and long flanking introns to be highly deregulated upon SFPQ knockdown. Moreover, SFPQ depletion leads to increased intron retention with concomitant induction of cryptic splicing, premature transcription termination, and polyadenylation, particularly prevalent for long introns. Aberrant splicing in the upstream and downstream regions of circRNA producing exons are critical for shaping the circRNAome, and specifically, we identify missplicing in the immediate upstream region to be a conserved driver of circRNA biogenesis. Collectively, our data show that SFPQ plays an important role in maintaining intron integrity by ensuring accurate splicing of long introns, and disclose novel features governing Alu-independent circRNA production.
Keywords: SFPQ; alternative splicing; chromosomes; circular RNA; gene expression; human; mouse; premature termination.
© 2021, Stagsted et al.
Conflict of interest statement
LS, EO, KE, TH No competing interests declared
Figures

References
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
