

Congenital Diseases of DNA Replication: Clinical Phenotypes and Molecular Mechanisms
- PMID: 33477564
- PMCID: PMC7831139
- DOI: 10.3390/ijms22020911
Congenital Diseases of DNA Replication: Clinical Phenotypes and Molecular Mechanisms
Abstract
Deoxyribonucleic acid (DNA) replication can be divided into three major steps: initiation, elongation and termination. Each time a human cell divides, these steps must be reiteratively carried out. Disruption of DNA replication can lead to genomic instability, with the accumulation of point mutations or larger chromosomal anomalies such as rearrangements. While cancer is the most common class of disease associated with genomic instability, several congenital diseases with dysfunctional DNA replication give rise to similar DNA alterations. In this review, we discuss all congenital diseases that arise from pathogenic variants in essential replication genes across the spectrum of aberrant replisome assembly, origin activation and DNA synthesis. For each of these conditions, we describe their clinical phenotypes as well as molecular studies aimed at determining the functional mechanisms of disease, including the assessment of genomic stability. By comparing and contrasting these diseases, we hope to illuminate how the disruption of DNA replication at distinct steps affects human health in a surprisingly cell-type-specific manner.
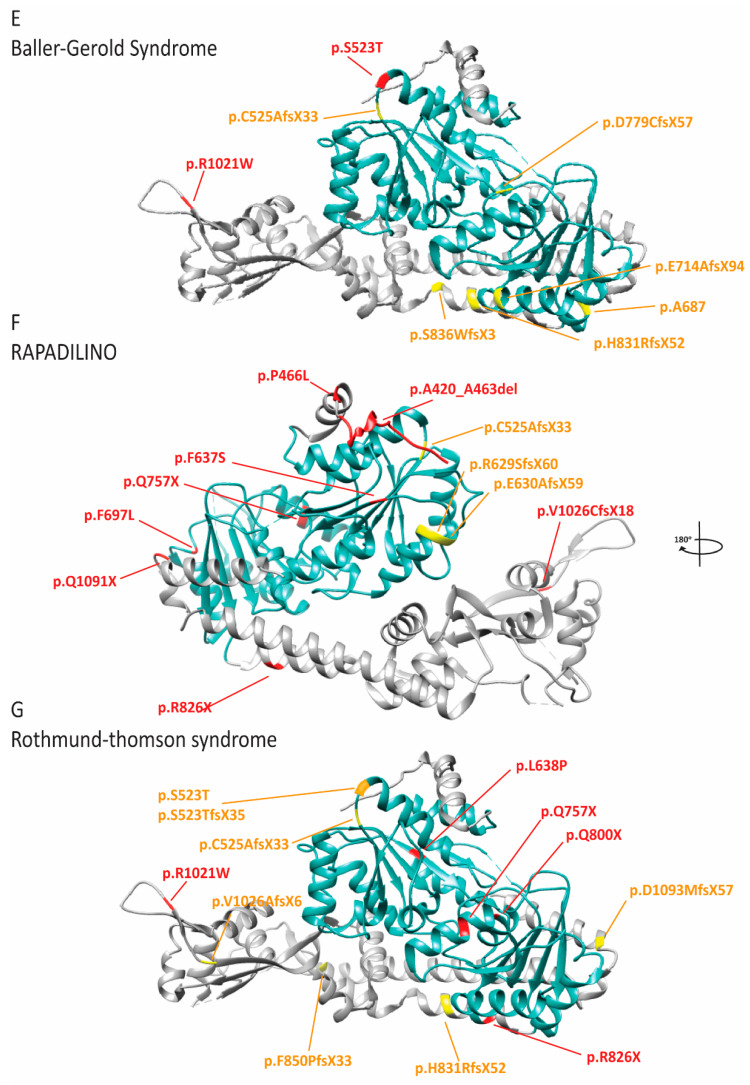
Keywords: Baller-Gerold syndrome; FILS syndrome; IMAGe syndrome; Meier-Gorlin syndrome; RAPADILINO; Rothmund-Thomson syndrome; Van Esch-O’Driscoll disease; X-linked pigmentary reticulate disorder; natural killer cell deficiency.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
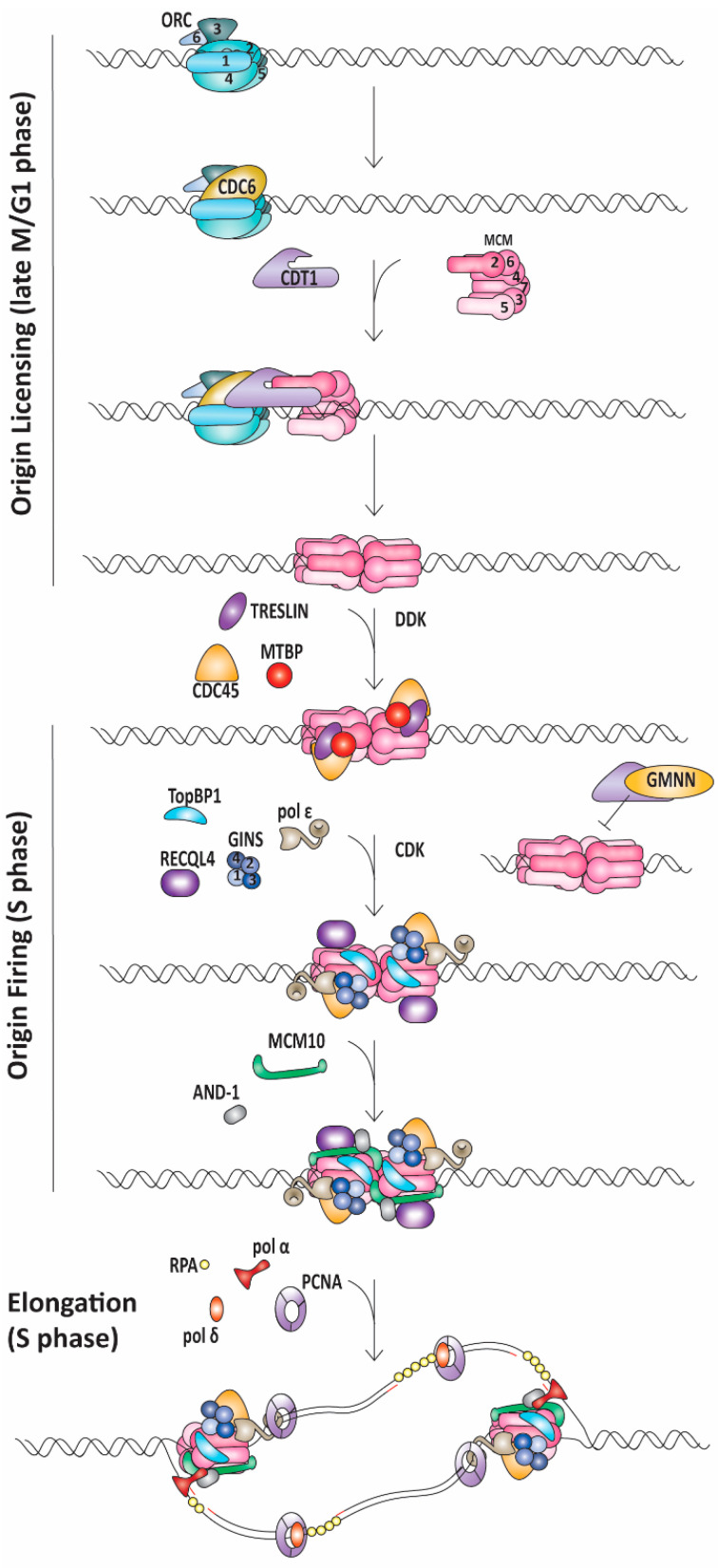
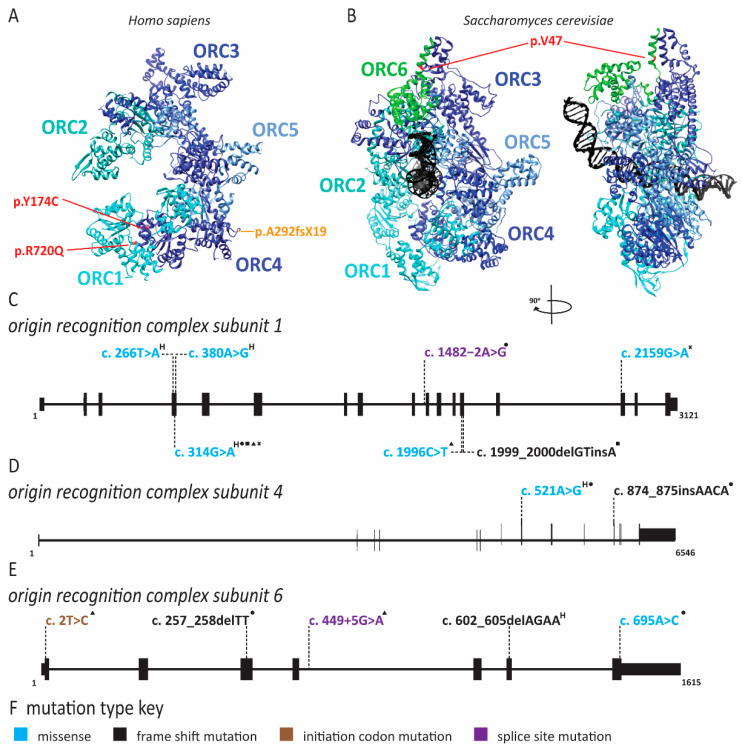
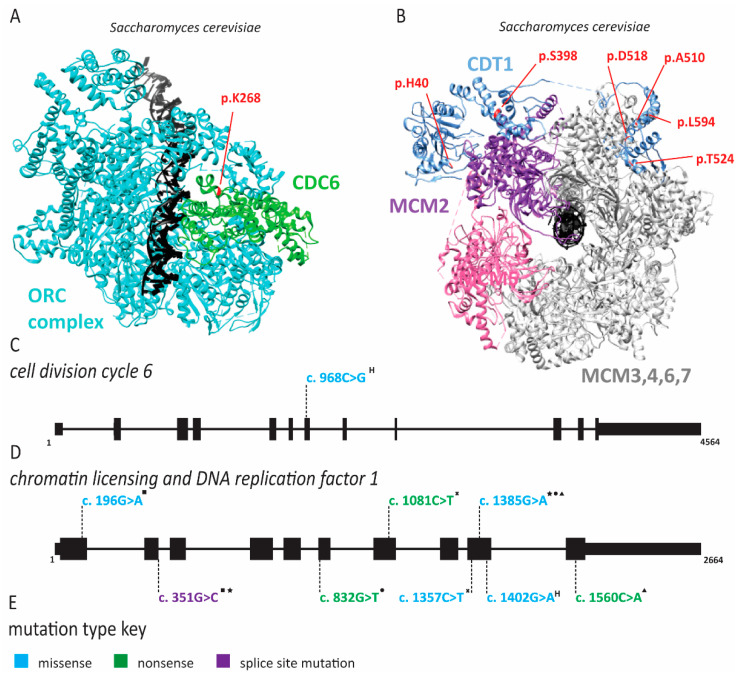
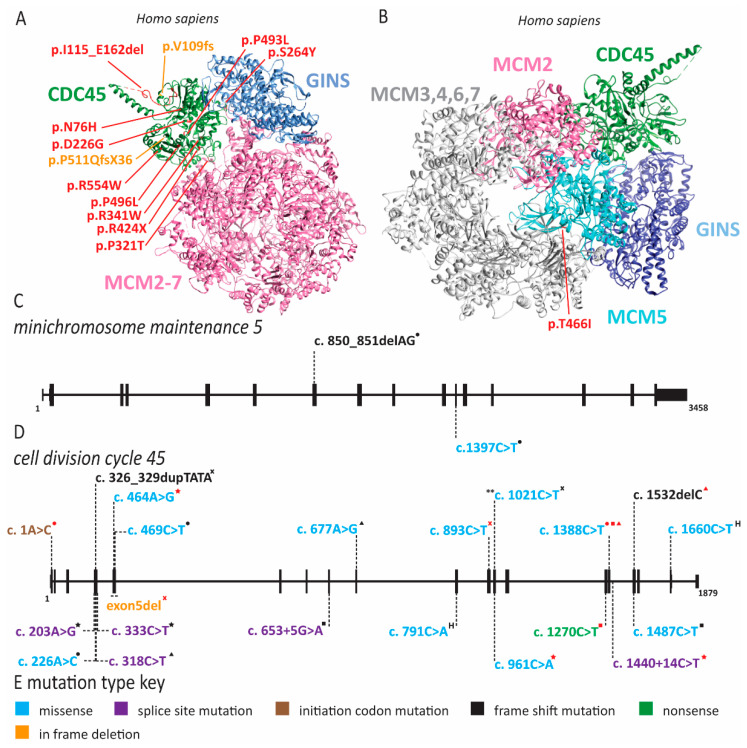
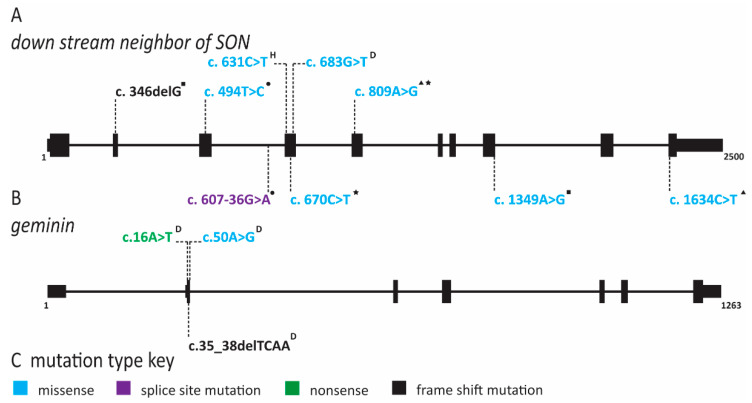
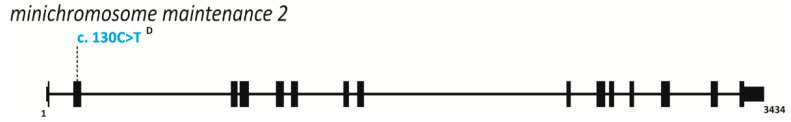
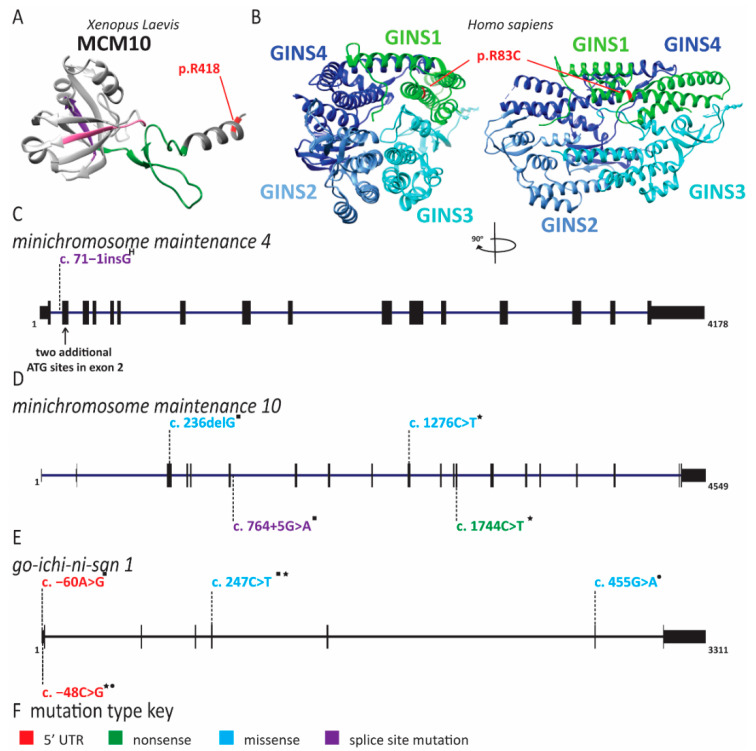
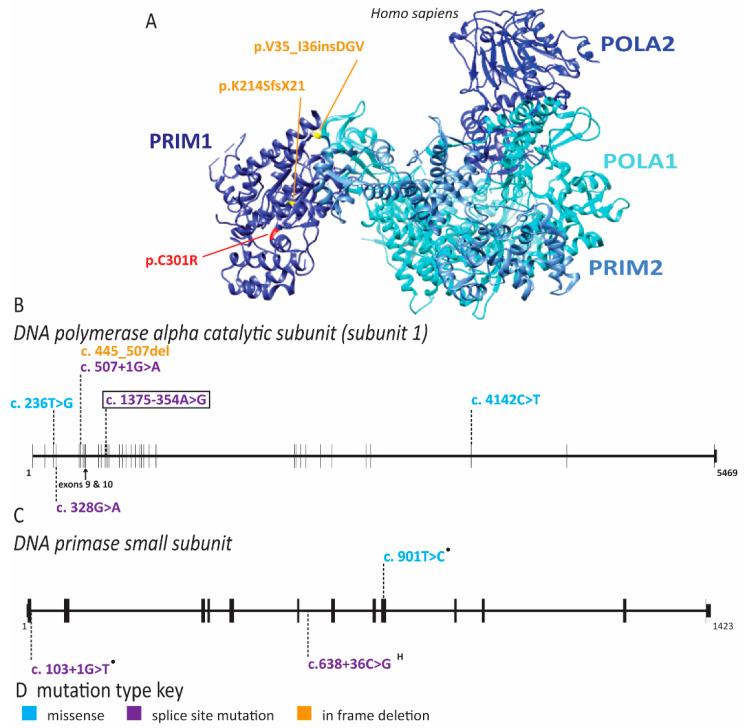
References
-
- Chandrasekaran S., Reidy T.K., Gowen J. Fundamental Aspects of DNA Replication. IntechOpen; London, UK: 2011. Regulation of DNA Replication Origin Licensing. - DOI
-
- Zhang J., Bellani M.A., James R.C., Pokharel D., Zhang Y., Reynolds J.J., McNee G.S., Jackson A.P., Stewart G.S., Seidman M.M. DONSON and FANCM associate with different replisomes distinguished by replication timing and chromatin domain. Nat. Commun. 2020;11:1–15. doi: 10.1038/s41467-020-17449-1. - DOI - PMC - PubMed
-
- Reynolds J.J., Bicknell L.S., Carroll P., Higgs M.R., Shaheen R., Murray J.E., Papadopoulos D.K., Leitch A., Murina O., Tarnauskaitė Ž., et al. Mutations in DONSON disrupt replication fork stability and cause microcephalic dwarfism. Nat. Genet. 2017;49:537–549. doi: 10.1038/ng.3790. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
