

Decoding the RNA viromes in rodent lungs provides new insight into the origin and evolutionary patterns of rodent-borne pathogens in Mainland Southeast Asia
- PMID: 33478588
- PMCID: PMC7818139
- DOI: 10.1186/s40168-020-00965-z
Decoding the RNA viromes in rodent lungs provides new insight into the origin and evolutionary patterns of rodent-borne pathogens in Mainland Southeast Asia
Abstract
Background: As the largest group of mammalian species, which are also widely distributed all over the world, rodents are the natural reservoirs for many diverse zoonotic viruses. A comprehensive understanding of the core virome of diverse rodents should therefore assist in efforts to reduce the risk of future emergence or re-emergence of rodent-borne zoonotic pathogens.
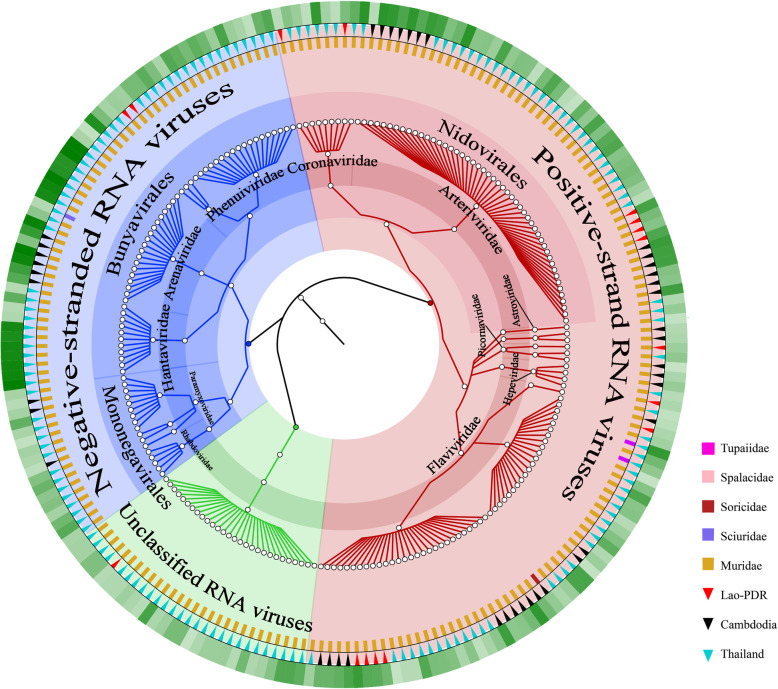
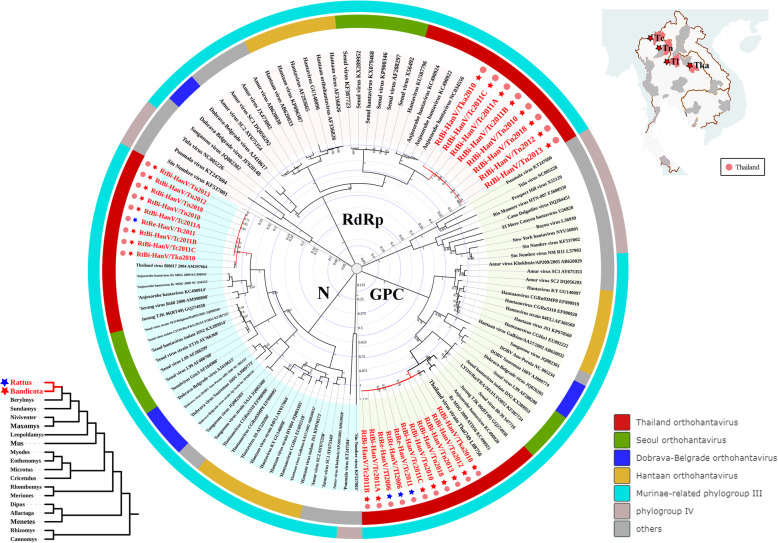
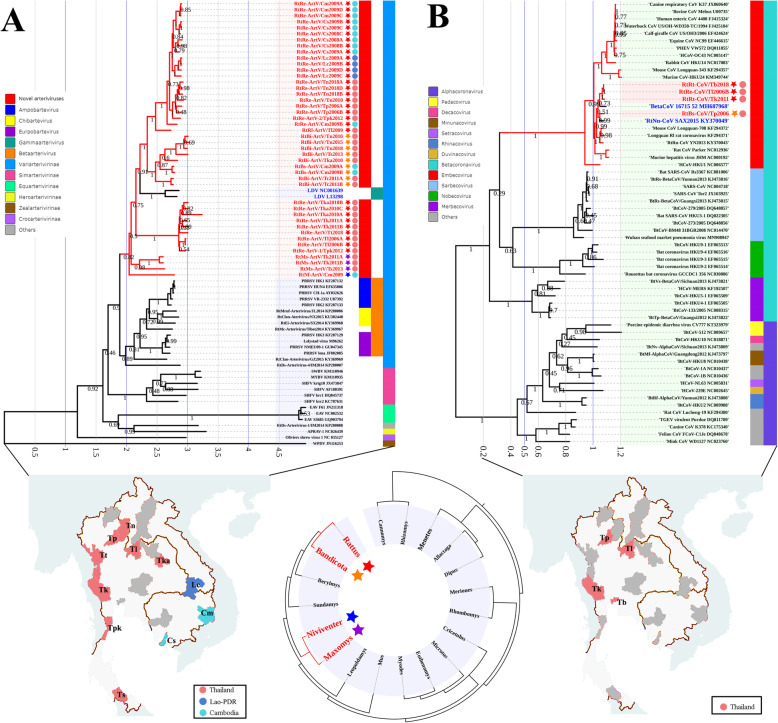
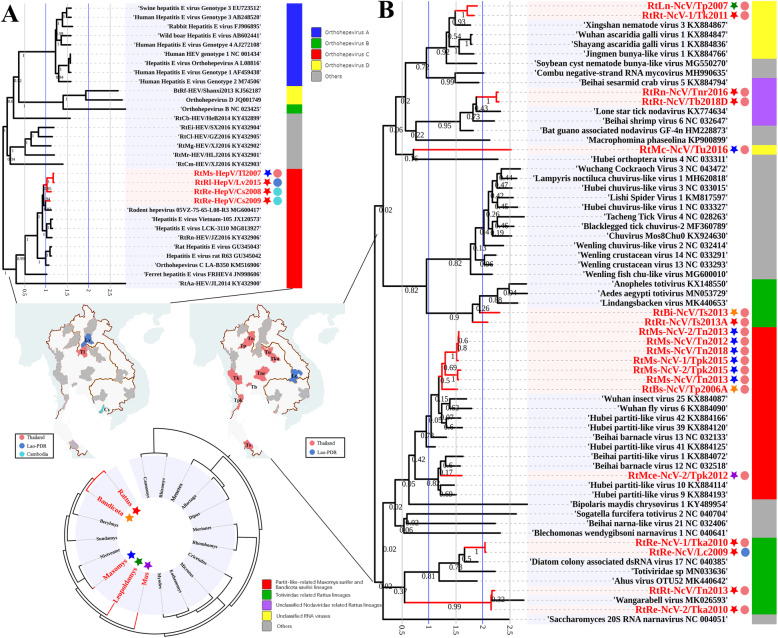
Results: This study aimed to describe the viral range that could be detected in the lungs of rodents from Mainland Southeast Asia. Lung samples were collected from 3284 rodents and insectivores of the orders Rodentia, Scandentia, and Eulipotyphla in eighteen provinces of Thailand, Lao PDR, and Cambodia throughout 2006-2018. Meta-transcriptomic analysis was used to outline the unique spectral characteristics of the mammalian viruses within these lungs and the ecological and genetic imprints of the novel viruses. Many mammalian- or arthropod-related viruses from distinct evolutionary lineages were reported for the first time in these species, and viruses related to known pathogens were characterized for their genomic and evolutionary characteristics, host species, and locations.
Conclusions: These results expand our understanding of the core viromes of rodents and insectivores from Mainland Southeast Asia and suggest that a high diversity of viruses remains to be found in rodent species of this area. These findings, combined with our previous virome data from China, increase our knowledge of the viral community in wildlife and arthropod vectors in emerging disease hotspots of East and Southeast Asia. Video abstract.
Keywords: Core virome; Emerging infectious diseases; Mainland Southeast Asia; Rodent lungs; Viral evolution.
Conflict of interest statement
The authors declare that they have no competing interests
Figures





References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
