

Noncanonical scaffolding of Gαi and β-arrestin by G protein-coupled receptors
- PMID: 33479120
- PMCID: PMC8005335
- DOI: 10.1126/science.aay1833
Noncanonical scaffolding of Gαi and β-arrestin by G protein-coupled receptors
Abstract
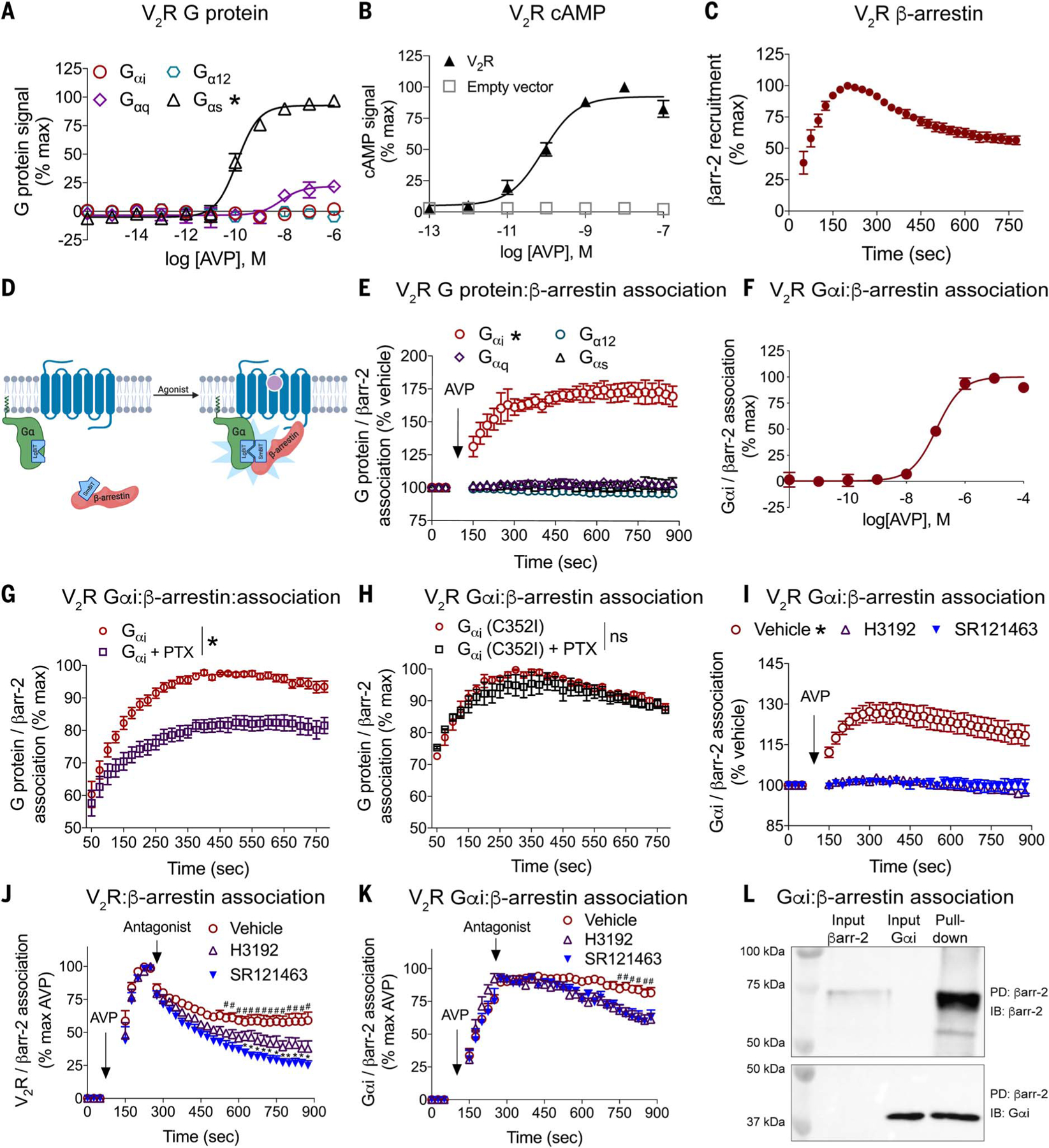
Heterotrimeric guanine nucleotide-binding protein (G protein)-coupled receptors (GPCRs) are common drug targets and canonically couple to specific Gα protein subtypes and β-arrestin adaptor proteins. G protein-mediated signaling and β-arrestin-mediated signaling have been considered separable. We show here that GPCRs promote a direct interaction between Gαi protein subtype family members and β-arrestins regardless of their canonical Gα protein subtype coupling. Gαi:β-arrestin complexes bound extracellular signal-regulated kinase (ERK), and their disruption impaired both ERK activation and cell migration, which is consistent with β-arrestins requiring a functional interaction with Gαi for certain signaling events. These results introduce a GPCR signaling mechanism distinct from canonical G protein activation in which GPCRs cause the formation of Gαi:β-arrestin signaling complexes.
Copyright © 2021 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Conflict of interest statement
Figures





References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
