

Further evidence supporting a potential role for ADH1B in obesity
- PMID: 33479282
- PMCID: PMC7820614
- DOI: 10.1038/s41598-020-80563-z
Further evidence supporting a potential role for ADH1B in obesity
Abstract
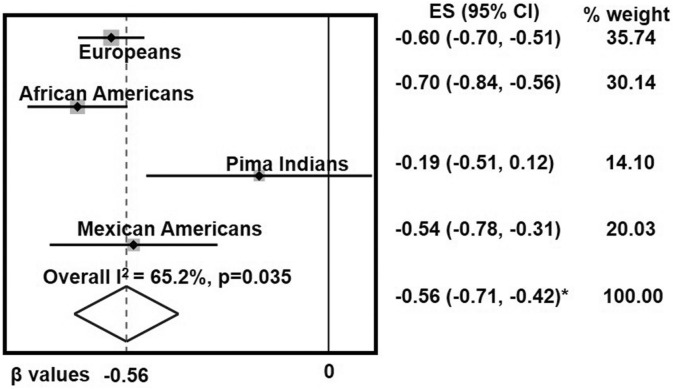
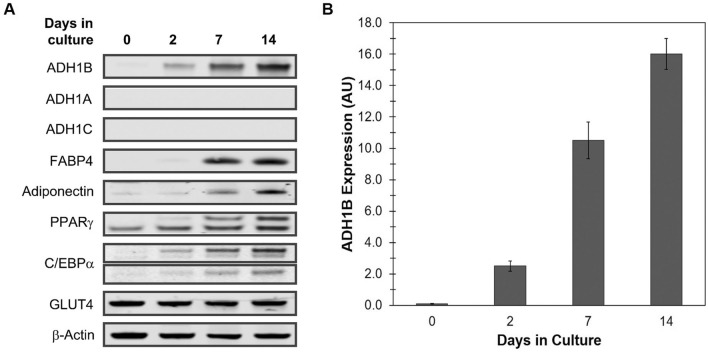
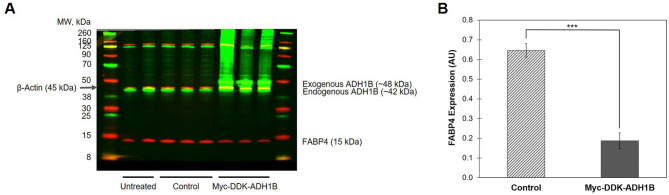
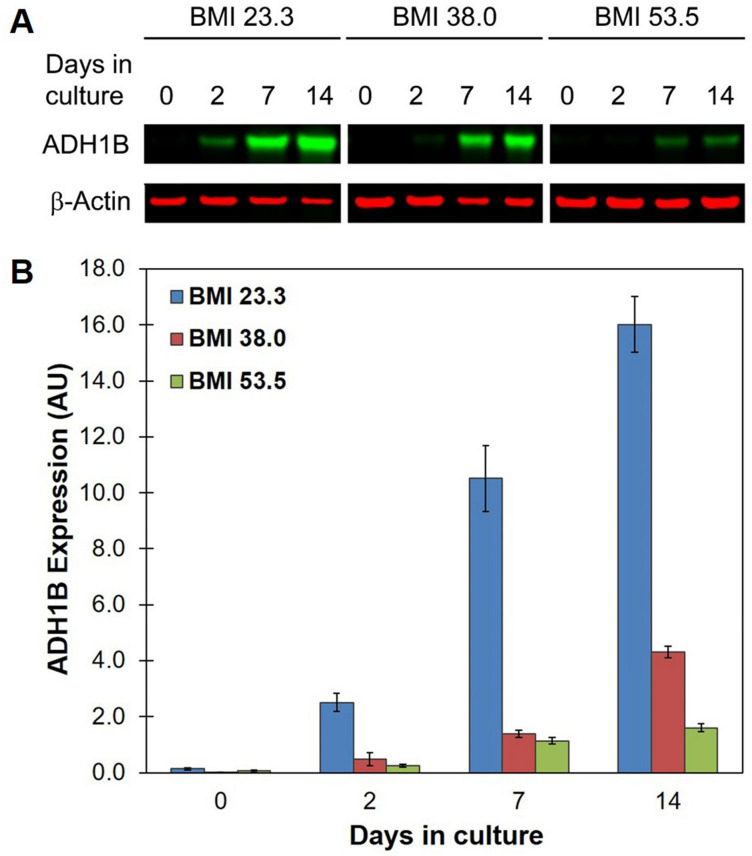
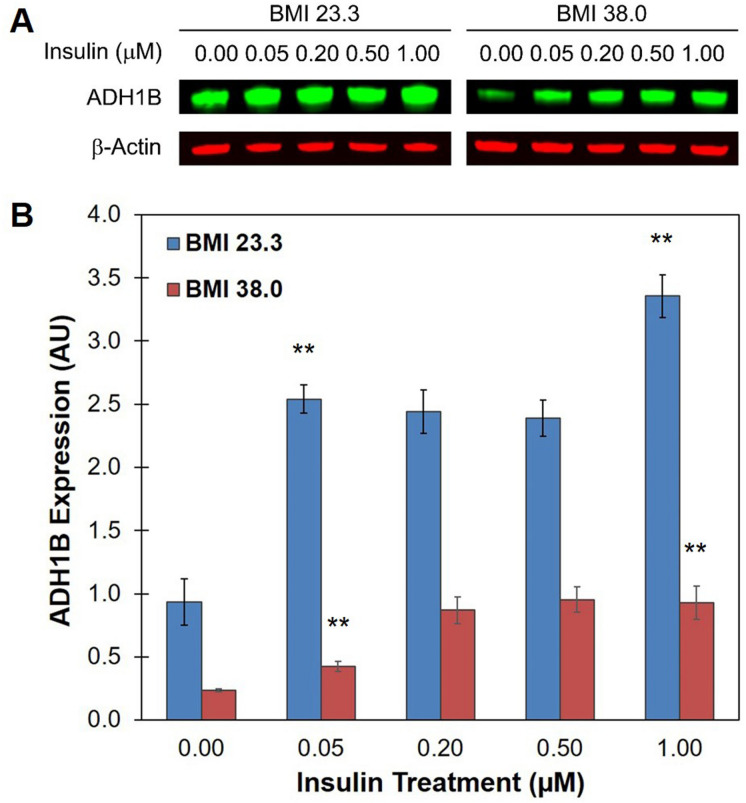
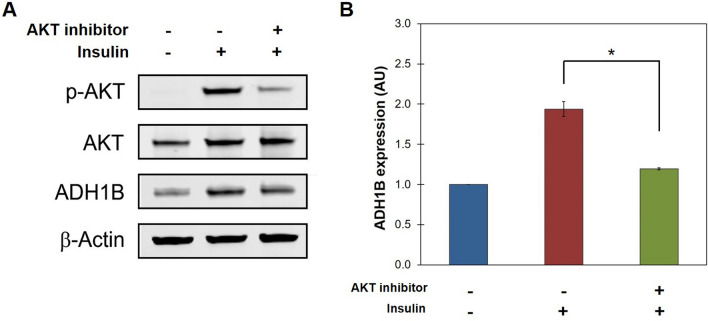
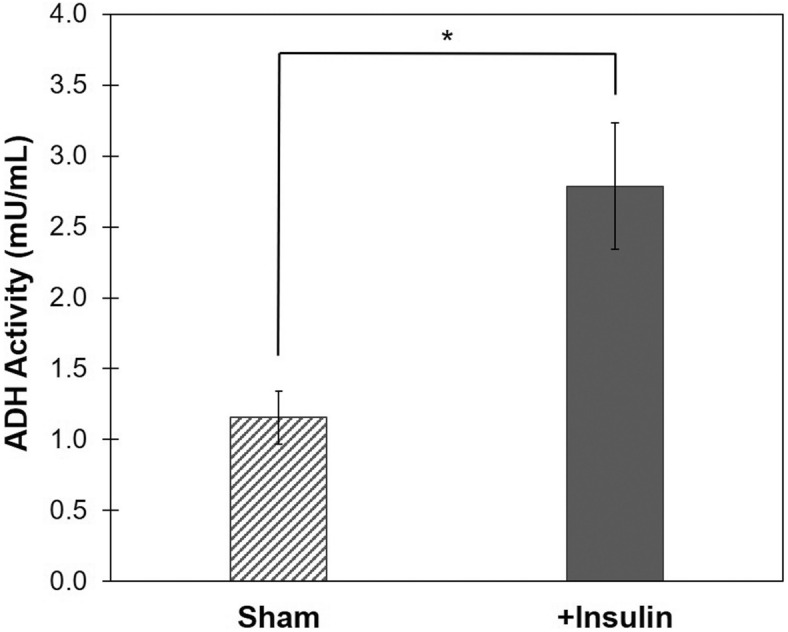
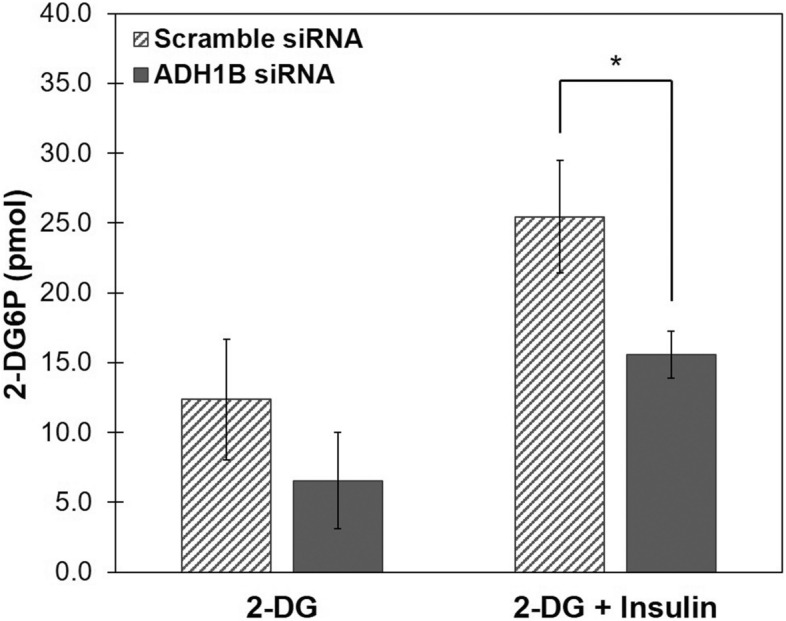
Insulin is an essential hormone that regulates glucose homeostasis and metabolism. Insulin resistance (IR) arises when tissues fail to respond to insulin, and it leads to serious health problems including Type 2 Diabetes (T2D). Obesity is a major contributor to the development of IR and T2D. We previously showed that gene expression of alcohol dehydrogenase 1B (ADH1B) was inversely correlated with obesity and IR in subcutaneous adipose tissue of Mexican Americans. In the current study, a meta-analysis of the relationship between ADH1B expression and BMI in Mexican Americans, African Americans, Europeans, and Pima Indians verified that BMI was increased with decreased ADH1B expression. Using established human subcutaneous pre-adipocyte cell lines derived from lean (BMI < 30 kg m-2) or obese (BMI ≥ 30 kg m-2) donors, we found that ADH1B protein expression increased substantially during differentiation, and overexpression of ADH1B inhibited fatty acid binding protein expression. Mature adipocytes from lean donors expressed ADH1B at higher levels than obese donors. Insulin further induced ADH1B protein expression as well as enzyme activity. Knockdown of ADH1B expression decreased insulin-stimulated glucose uptake. Our findings suggest that ADH1B is involved in the proper development and metabolic activity of adipose tissues and this function is suppressed by obesity.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- US DHHS, CDC . Chronic Diseases and Their Risk factors: The Nation's Leading Causes of Death. Atlanta: US DHHS, CDC; 1999.
-
- Centers_for_Disease_Control_and_Prevention (U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, Atlanta, 2011) http://www.cdc.gov/chronicdisease/resources/publications/aag/ddt.htm.
-
- DeFronzo R. Pathogenesis of type 2 diabetes: Metabolic and molecular implications of identifying diabetes genes. Diabetes Rev. 1997;5:177–269.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
