

p53 is a central regulator driving neurodegeneration caused by C9orf72 poly(PR)
- PMID: 33482083
- PMCID: PMC7886018
- DOI: 10.1016/j.cell.2020.12.025
p53 is a central regulator driving neurodegeneration caused by C9orf72 poly(PR)
Abstract
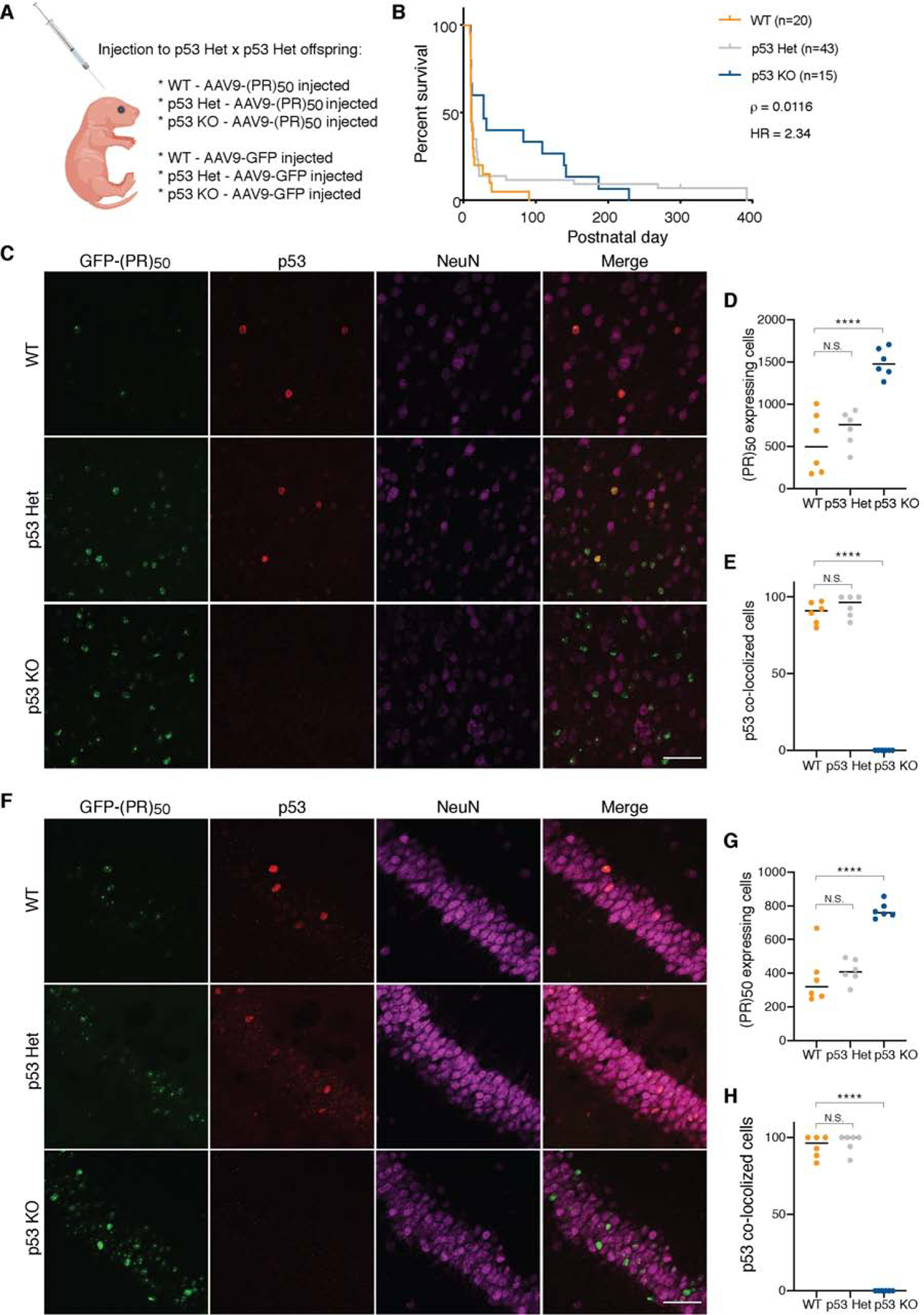
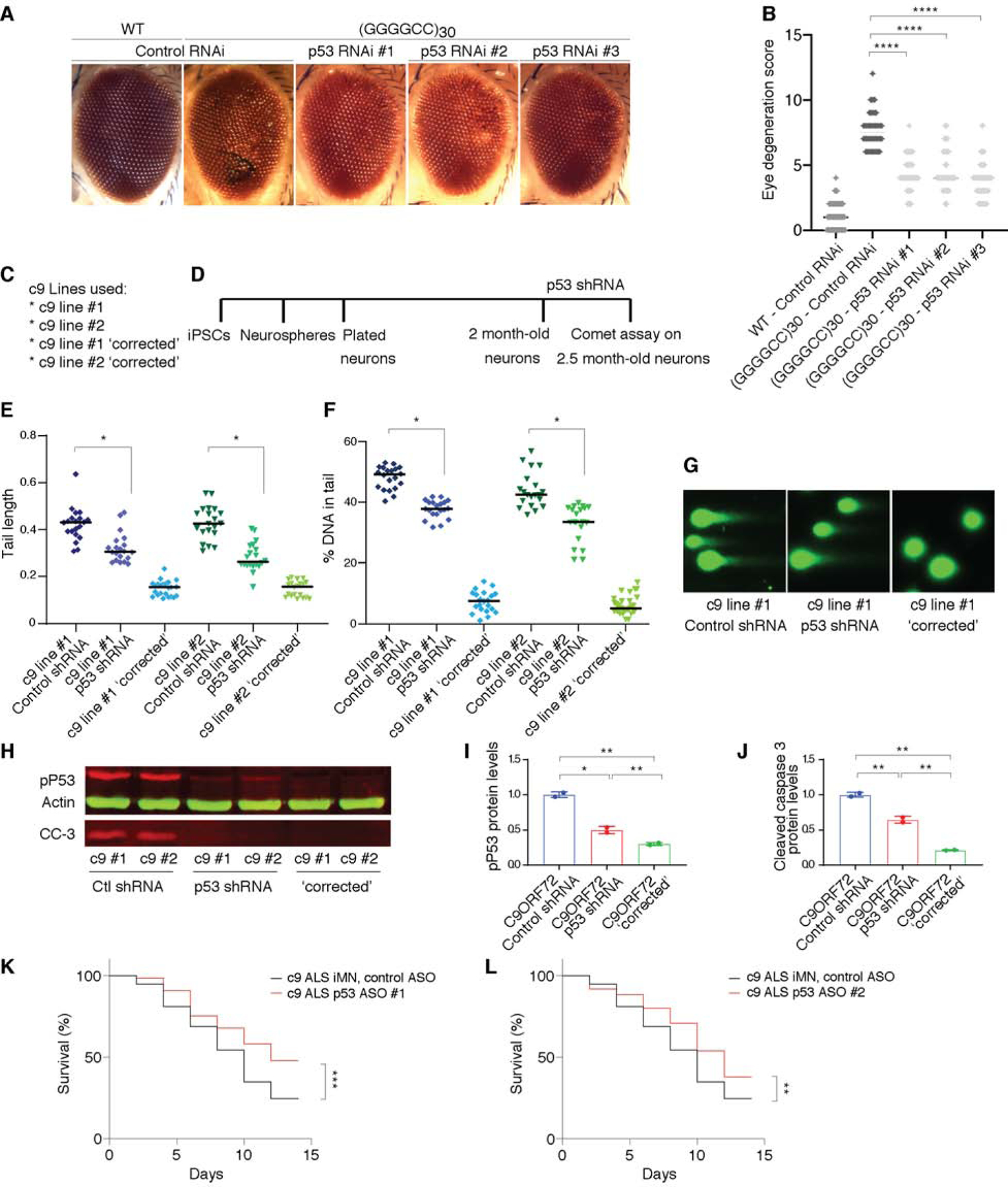
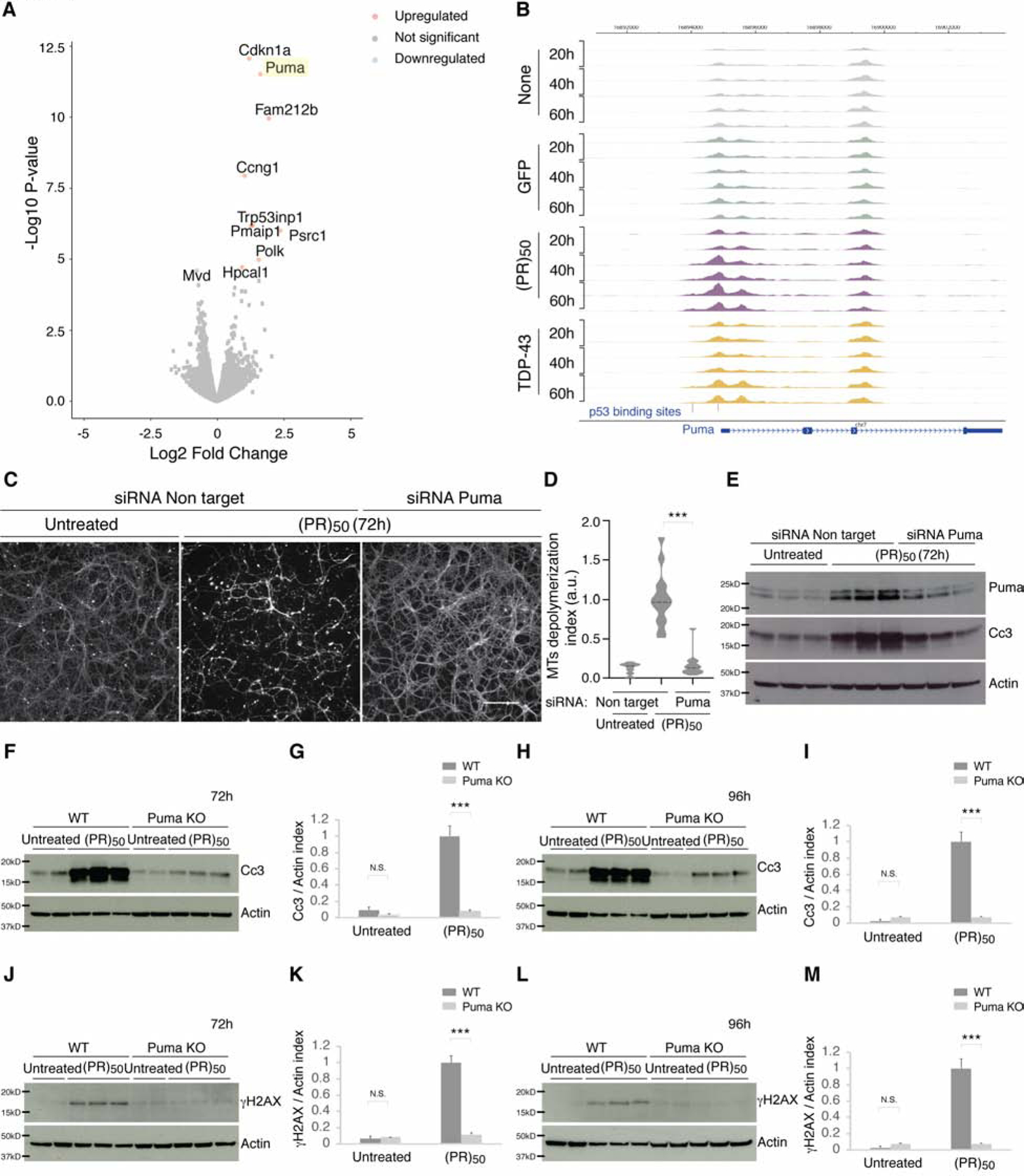
The most common genetic cause of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) is a GGGGCC repeat expansion in the C9orf72 gene. We developed a platform to interrogate the chromatin accessibility landscape and transcriptional program within neurons during degeneration. We provide evidence that neurons expressing the dipeptide repeat protein poly(proline-arginine), translated from the C9orf72 repeat expansion, activate a highly specific transcriptional program, exemplified by a single transcription factor, p53. Ablating p53 in mice completely rescued neurons from degeneration and markedly increased survival in a C9orf72 mouse model. p53 reduction also rescued axonal degeneration caused by poly(glycine-arginine), increased survival of C9orf72 ALS/FTD-patient-induced pluripotent stem cell (iPSC)-derived motor neurons, and mitigated neurodegeneration in a C9orf72 fly model. We show that p53 activates a downstream transcriptional program, including Puma, which drives neurodegeneration. These data demonstrate a neurodegenerative mechanism dynamically regulated through transcription-factor-binding events and provide a framework to apply chromatin accessibility and transcription program profiles to neurodegeneration.
Keywords: ATAC-seq; C9orf72; TDP-43; amyotrophic lateral sclerosis; axonal degeneration; neurodegeneration; p53; puma.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests A.D.G. has served as a consultant for Aquinnah Pharmaceuticals, Prevail Therapeutics, and Third Rock Ventures and is a scientific founder of Maze Therapeutics. W.J.G. has affiliations with 10x Genomics (consultant), Guardant Health (consultant), and Protillion Biosciences (co-founder and consultant). J.K.I. is a co-founder of Acurastem. D.H.G has served as a consultant for Acurastem, Axial Biosciences, and Roche
Figures
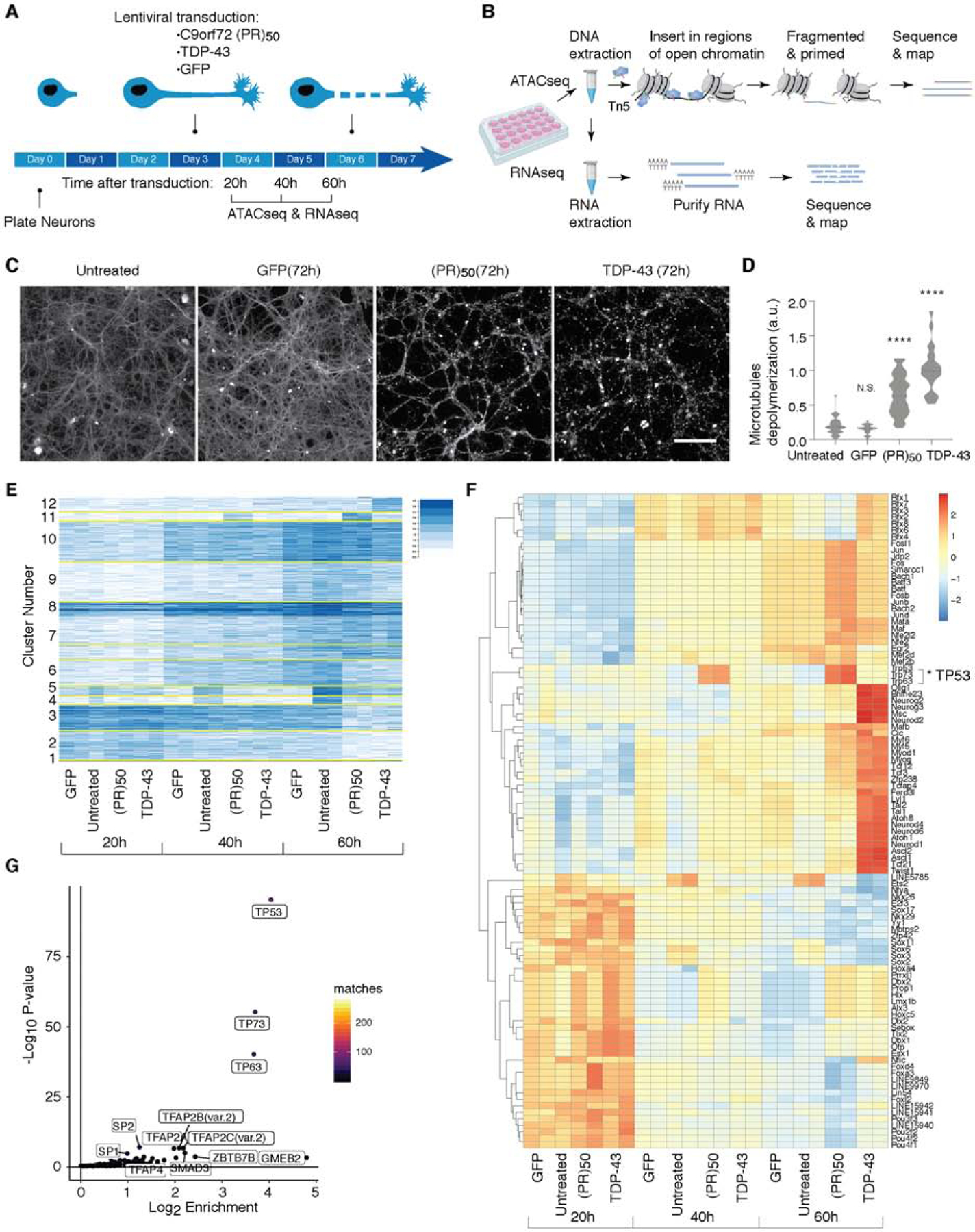
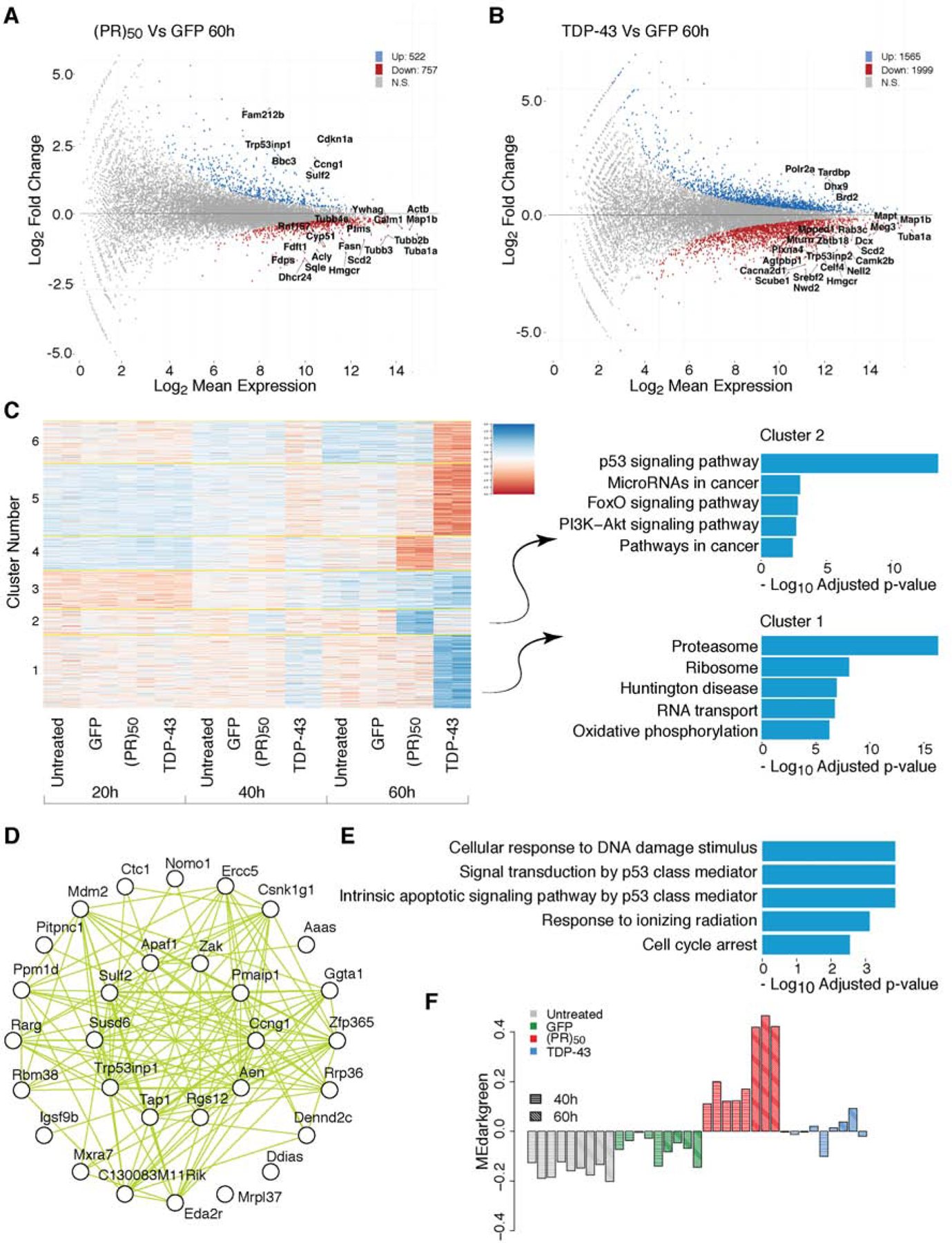
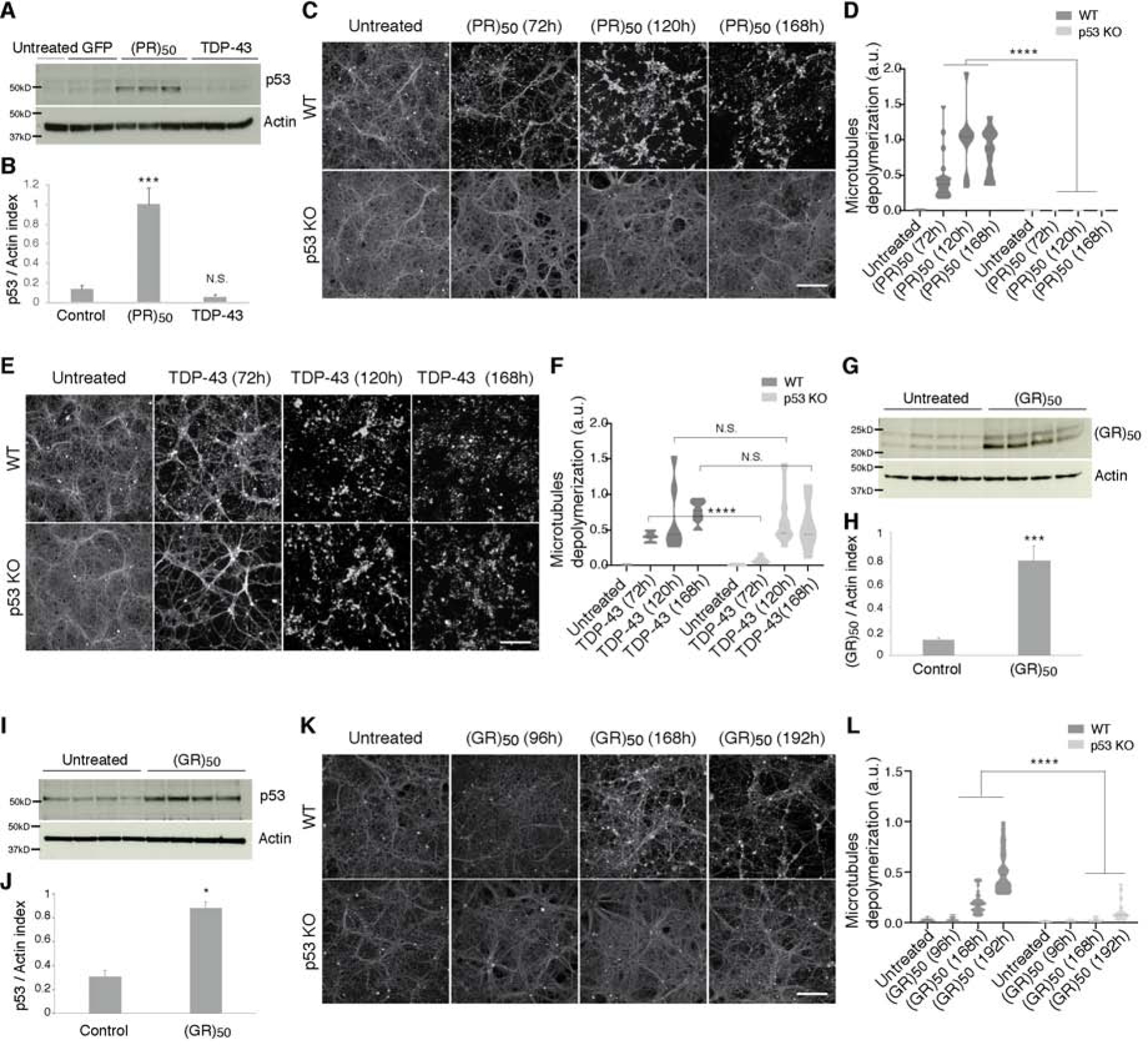
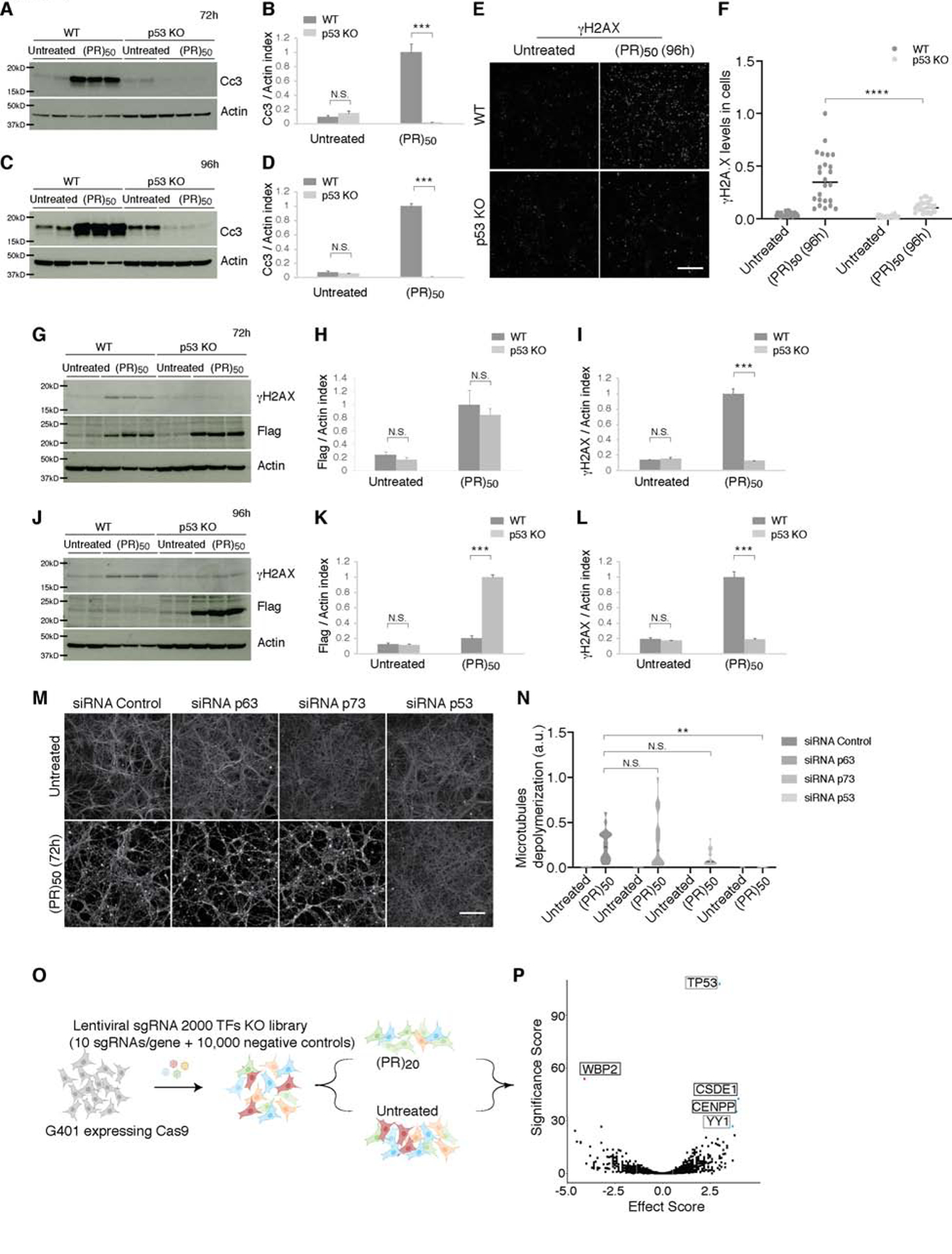
Comment in
-
Open Access: A Role for p53 in c9ALS/FTD?Trends Genet. 2021 May;37(5):404-406. doi: 10.1016/j.tig.2021.01.008. Epub 2021 Feb 4. Trends Genet. 2021. PMID: 33551183
References
-
- Akizuki M, Yamashita H, Uemura K, Maruyama H, Kawakami H, Ito H, and Takahashi R (2013). Optineurin suppression causes neuronal cell death via NF-kappaB pathway. J Neurochem 126, 699–704. - PubMed
-
- Bae BI, Xu H, Igarashi S, Fujimuro M, Agrawal N, Taya Y, Hayward SD, Moran TH, Montell C, Ross CA, et al. (2005). p53 mediates cellular dysfunction and behavioral abnormalities in Huntington’s disease. Neuron 47, 29–41. - PubMed
-
- Barbosa LF, Cerqueira FM, Macedo AF, Garcia CC, Angeli JP, Schumacher RI, Sogayar MC, Augusto O, Carri MT, Di Mascio P, et al. (2010). Increased SOD1 association with chromatin, DNA damage, p53 activation, and apoptosis in a cellular model of SOD1-linked ALS. Biochim Biophys Acta 1802, 462–471. - PubMed
-
- Barry S, Baird G, Lascelles K, Bunton P, and Hedderly T (2011). Neurodevelopmental movement disorders - an update on childhood motor stereotypies. Dev Med Child Neurol 53, 979–985. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R35 NS097273/NS/NINDS NIH HHS/United States
- RF1 NS101986/NS/NINDS NIH HHS/United States
- K01 AG049152/AG/NIA NIH HHS/United States
- P30 NS069375/NS/NINDS NIH HHS/United States
- U19 AI057266/AI/NIAID NIH HHS/United States
- R01 NS097850/NS/NINDS NIH HHS/United States
- R35 CA197591/CA/NCI NIH HHS/United States
- S10 OD020141/OD/NIH HHS/United States
- P01 AG019724/AG/NIA NIH HHS/United States
- P50 HG007735/HG/NHGRI NIH HHS/United States
- P01 NS084974/NS/NINDS NIH HHS/United States
- UM1 HG009436/HG/NHGRI NIH HHS/United States
- R01 MH109912/MH/NIMH NIH HHS/United States
- R01 NS094239/NS/NINDS NIH HHS/United States
- RM1 HG007735/HG/NHGRI NIH HHS/United States
- R01 HG008140/HG/NHGRI NIH HHS/United States
- R01 AG062588/AG/NIA NIH HHS/United States
- R37 NS057553/NS/NINDS NIH HHS/United States
- R01 NS057553/NS/NINDS NIH HHS/United States
- R01 NS089786/NS/NINDS NIH HHS/United States
- R01 DC015530/DC/NIDCD NIH HHS/United States
- RF1 AG064690/AG/NIA NIH HHS/United States
- P01 NS099114/NS/NINDS NIH HHS/United States
- UM1 HG009442/HG/NHGRI NIH HHS/United States
- T32 DC009975/DC/NIDCD NIH HHS/United States
- R01 NS101986/NS/NINDS NIH HHS/United States
- F32 MH114620/MH/NIMH NIH HHS/United States
- R35 NS097263/NS/NINDS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
