

Molecular mechanisms of telomere biology disorders
- PMID: 33482595
- PMCID: PMC7948428
- DOI: 10.1074/jbc.REV120.014017
Molecular mechanisms of telomere biology disorders
Abstract
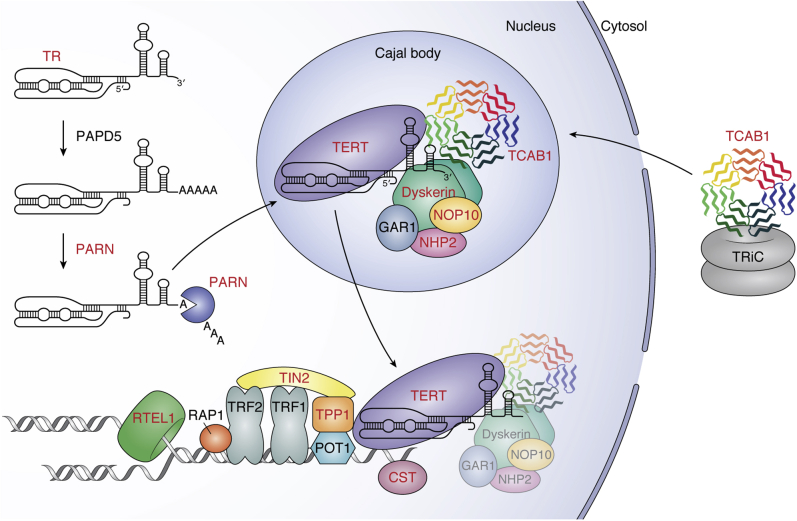
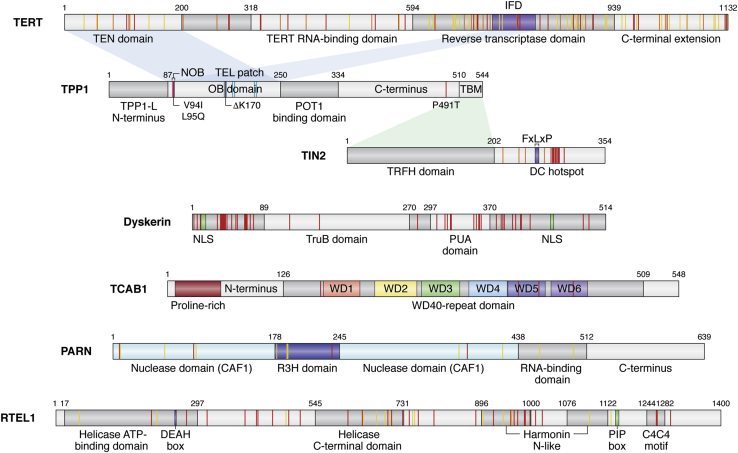
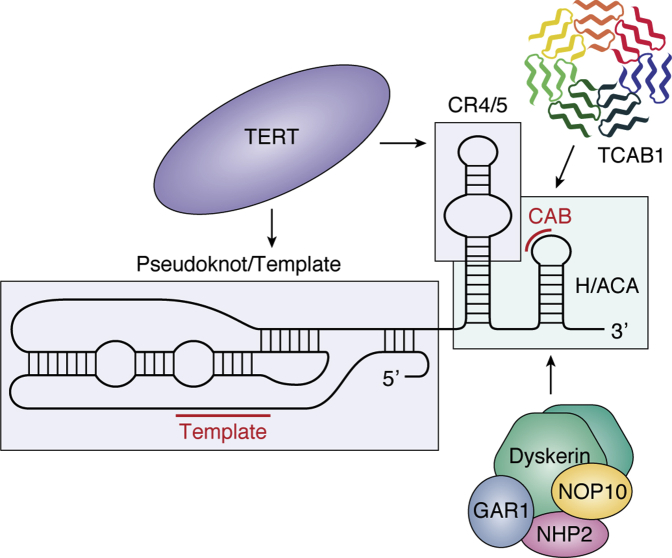
Genetic mutations that affect telomerase function or telomere maintenance result in a variety of diseases collectively called telomeropathies. This wide spectrum of disorders, which include dyskeratosis congenita, pulmonary fibrosis, and aplastic anemia, is characterized by severely short telomeres, often resulting in hematopoietic stem cell failure in the most severe cases. Recent work has focused on understanding the molecular basis of these diseases. Mutations in the catalytic TERT and TR subunits of telomerase compromise activity, while others, such as those found in the telomeric protein TPP1, reduce the recruitment of telomerase to the telomere. Mutant telomerase-associated proteins TCAB1 and dyskerin and the telomerase RNA maturation component poly(A)-specific ribonuclease affect the maturation and stability of telomerase. In contrast, disease-associated mutations in either CTC1 or RTEL1 are more broadly associated with telomere replication defects. Yet even with the recent surge in studies decoding the mechanisms underlying these diseases, a significant proportion of dyskeratosis congenita mutations remain uncharacterized or poorly understood. Here we review the current understanding of the molecular basis of telomeropathies and highlight experimental data that illustrate how genetic mutations drive telomere shortening and dysfunction in these patients. This review connects insights from both clinical and molecular studies to create a comprehensive view of the underlying mechanisms that drive these diseases. Through this, we emphasize recent advances in therapeutics and pinpoint disease-associated variants that remain poorly defined in their mechanism of action. Finally, we suggest future avenues of research that will deepen our understanding of telomere biology and telomere-related disease.
Keywords: dyskeratosis congenita; telomerase; telomerase assembly; telomerase recruitment; telomere; telomere shortening; telomeropathies.
Copyright © 2020 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest The authors declare that they have no conflicts of interest with the contents of this article.
Figures
References
-
- Palm W., de Lange T. How shelterin protects mammalian telomeres. Annu. Rev. Genet. 2008;42:301–334. - PubMed
-
- Greider C.W., Blackburn E.H. The telomere terminal transferase of tetrahymena is a ribonucleoprotein enzyme with two kinds of primer specificity. Cell. 1987;51:887–898. - PubMed
-
- Blackburn E.H., Greider C.W., Henderson E., Lee M.S., Shampay J., Shippen-Lentz D. Recognition and elongation of telomeres by telomerase. Genome. 1989;31:553–560. - PubMed
-
- Lingner J., Hughes T.R., Shevchenko A., Mann M., Lundblad V., Cech T.R. Reverse transcriptase motifs in the catalytic subunit of telomerase. Science. 1997;276:561–567. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
