

A master protocol to investigate a novel therapy acetyl-L-leucine for three ultra-rare neurodegenerative diseases: Niemann-Pick type C, the GM2 gangliosidoses, and ataxia telangiectasia
- PMID: 33482890
- PMCID: PMC7821839
- DOI: 10.1186/s13063-020-05009-3
A master protocol to investigate a novel therapy acetyl-L-leucine for three ultra-rare neurodegenerative diseases: Niemann-Pick type C, the GM2 gangliosidoses, and ataxia telangiectasia
Abstract
Background: The lack of approved treatments for the majority of rare diseases is reflective of the unique challenges of orphan drug development. Novel methodologies, including new functionally relevant endpoints, are needed to render the development process more feasible and appropriate for these rare populations and thereby expedite the approval of promising treatments to address patients' high unmet medical need. Here, we describe the development of an innovative master protocol and primary outcome assessment to investigate the modified amino acid N-acetyl-L-leucine (Sponsor Code: IB1001) in three separate, multinational, phase II trials for three ultra-rare, autosomal-recessive, neurodegenerative disorders: Niemann-Pick disease type C (NPC), GM2 gangliosidoses (Tay-Sachs and Sandhoff disease; "GM2"), and ataxia telangiectasia (A-T).
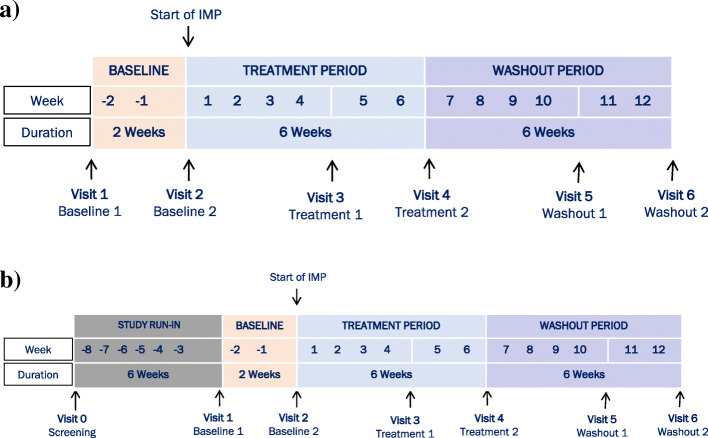
Methods/design: The innovative IB1001 master protocol and novel CI-CS primary endpoints were developed through a close collaboration between the Industry Sponsor, Key Opinion Leaders, representatives of the Patient Communities, and National Regulatory Authorities. As a result, the open-label, rater-blinded study design is considerate of the practical limitations of recruitment and retention of subjects in these ultra-orphan populations. The novel primary endpoint, the Clinical Impression of Change in Severity© (CI-CS), accommodates the heterogenous clinical presentation of NPC, GM2, and A-T: at screening, the principal investigator appoints for each patient a primary anchor test (either the 8-m walk test (8MWT) or 9-hole peg test of the dominant hand (9HPT-D)) based on his/her unique clinical symptoms. The anchor tests are videoed in a standardized manner at each visit to capture all aspects related to the patient's functional performance. The CI-CS assessment is ultimately performed by independent, blinded raters who compare videos of the primary anchor test from three periods: baseline, the end of treatment, and the end of a post-treatment washout. Blinded to the time point of each video, the raters make an objective comparison scored on a 7-point Likert scale of the change in the severity of the patient's neurological signs and symptoms from video A to video B. To investigate both the symptomatic and disease-modifying effects of treatment, N-acetyl-L-leucine is assessed during two treatment sequences: a 6-week parent study and 1-year extension phase.
Discussion: The novel CI-CS assessment, developed through a collaboration of all stakeholders, is advantageous in that it better ensures the primary endpoint is functionally relevant for each patient, is able to capture small but meaningful clinical changes critical to the patients' quality of life (fine-motor skills; gait), and blinds the primary outcome assessment. The results of these three trials will inform whether N-acetyl-L-leucine is an effective treatment for NPC, GM2, and A-T and can also serve as a new therapeutic paradigm for the development of future treatments for other orphan diseases.
Trial registration: The three trials (IB1001-201 for Niemann-Pick disease type C (NPC), IB1001-202 for GM2 gangliosidoses (Tay-Sachs and Sandhoff), IB1001-203 for ataxia telangiectasia (A-T)) have been registered at www.clinicaltrials.gov (NCT03759639; NCT03759665; NCT03759678), www.clinicaltrialsregister.eu (EudraCT: 2018-004331-71; 2018-004406-25; 2018-004407-39), and https://www.germanctr.de (DR KS-ID: DRKS00016567; DRKS00017539; DRKS00020511).
Keywords: Ataxia telangiectasia; Cerebellar ataxia; GM2 gangliosidosis; Lysosomal storage disease; N-acetyl-L-leucine; Niemann-Pick disease type C (NPC); Pharmaceutical intervention; Sandhoff disease; Single-blinded trial; Symptomatic treatment; Tay-Sachs disease (TSD).
Conflict of interest statement
MF is a co-founder, shareholder, and Chairman of IntraBio. TF and IB are officers and shareholders of IntraBio. FP is a cofounder, shareholder, and consultant to IntraBio and consultant to Actelion and Orphazyme. MS is a shareholder to IntraBio, and consultant for Abbott, Actelion, AurisMedical, Heel, IntraBio and Sensorion; he has received speaker’s honoraria from Abbott, Actelion, Auris Medical, Biogen, Eisai, Grünenthal, GSK, Henning Pharma, Interacoustics, Johnson & Johnson, MSD, Otometrics, Pierre-Fabre, TEVA, UCB. He is the distributor of M-glasses and positional vertigo App. AG and GC are cofounders, shareholders, and consultants to IntraBio. MP is a shareholder of IntraBio and has received consulting fees, honoraria, and research grants from Actelion Pharmaceuticals Ltd. and Biomarin. CV and SF are consultants and shareholders of IntraBio. UG, TM, GB, and RKay are consultants to IntraBio. TBE received honoraria for lecturing from Actelion and Sanofi Genzyme.
IntraBio owns patents EP3359146 and EP3416631 (related to treatment of lysosomal storage disorders and neurodegenerative diseases with acetyl-leucine and its analogues).
IntraBio has pending patent applications EP19174007.5, EP3482754, PCT/US2018/056420, PCT/US2018/018420, PCT/IB2018/054676, PCT/IB2019/051214, PCT/IB2017/054928, PCT/GB2017/051090, PCT/IB2017/054929, USPTO 62/812,987, USPTO 62/842,296, USPTO 62/888,894, USPTO 62/895,144, USPTO 62/868,383, USPTO 62/931,003, USPTO 62/960,637, and PCT/IB2019/060525 relating to treatment of lysosomal storage disorders, neurodegenerative diseases, and neurodegeneration with acetyl-leucine and its analogues.
Figures

References
-
- Public Law. Orphan Drug Act of 1983. Pub L. No. 97–414, 96 Stat. 2049. Available from: https://www.fda.gov/media/99546/download. [cited 2020 Feb 25].
-
- RARE Facts [Internet]. Glob. Genes. [cited 2020 Feb 25]. Available from: https://globalgenes.org/rare-facts/.
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
