

Integrative computational approach identifies drug targets in CD4+ T-cell-mediated immune disorders
- PMID: 33483502
- PMCID: PMC7822845
- DOI: 10.1038/s41540-020-00165-3
Integrative computational approach identifies drug targets in CD4+ T-cell-mediated immune disorders
Abstract
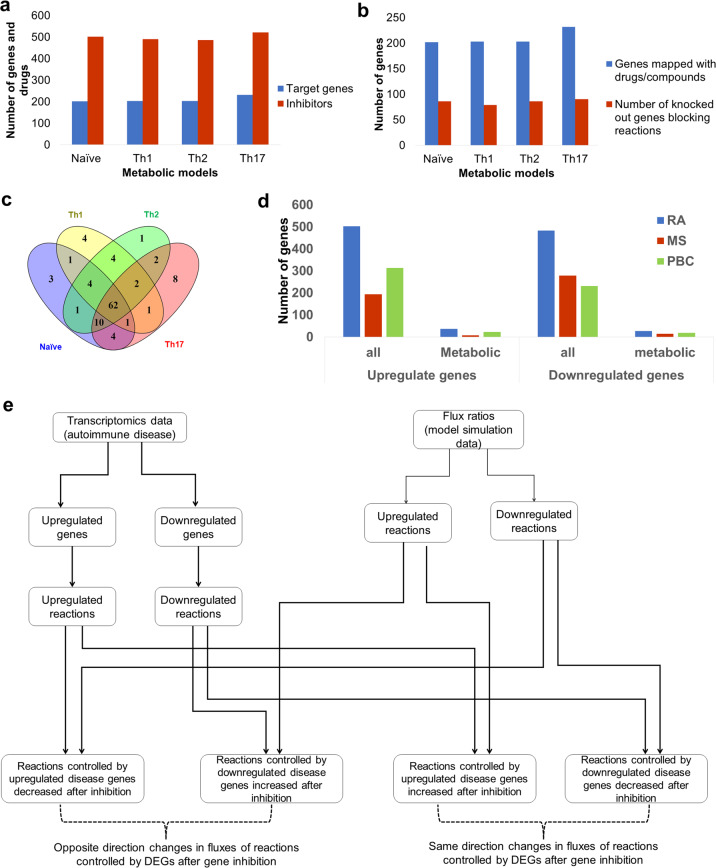
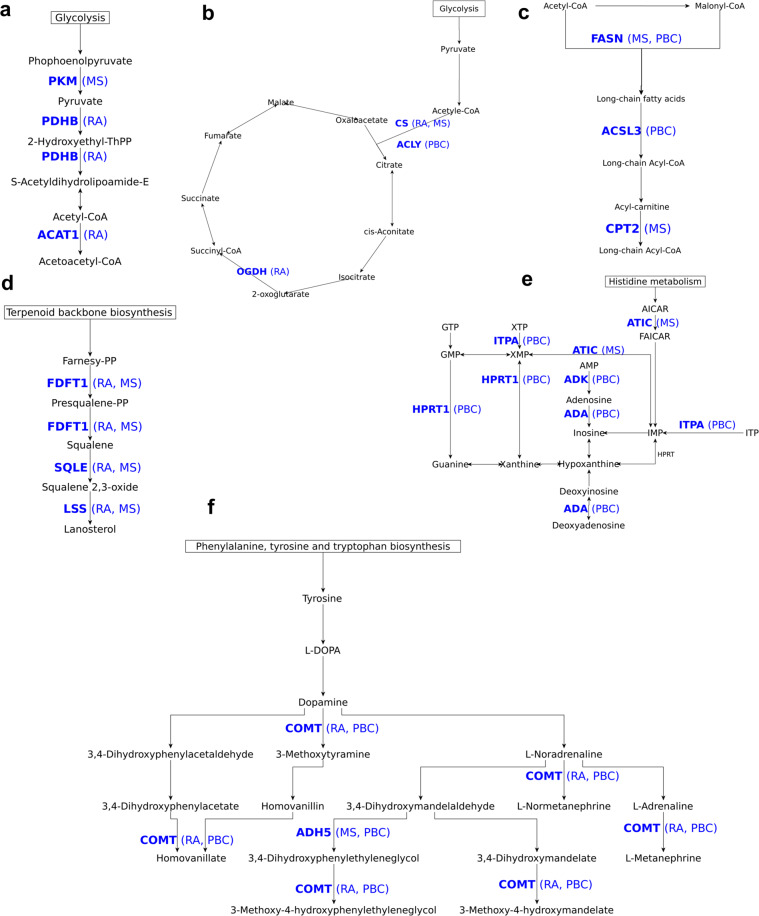
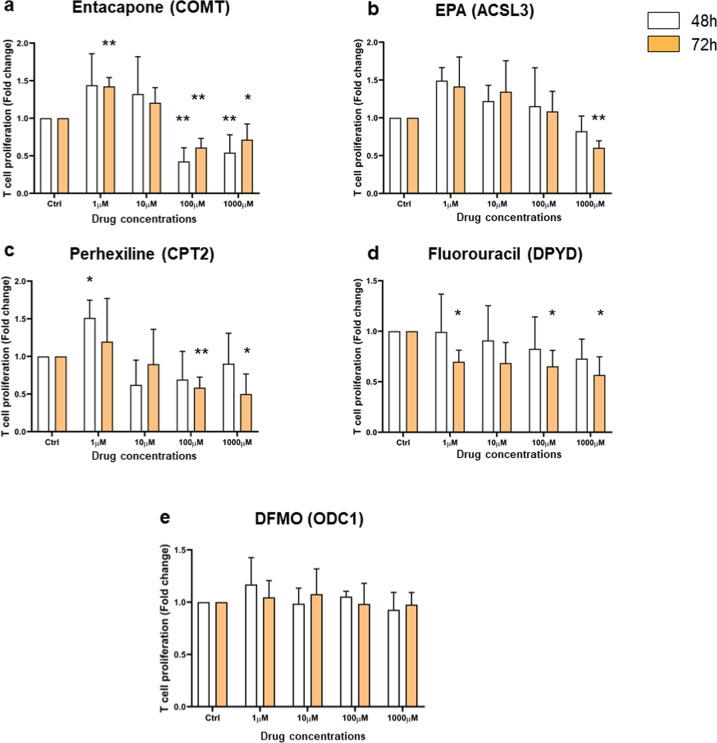
CD4+ T cells provide adaptive immunity against pathogens and abnormal cells, and they are also associated with various immune-related diseases. CD4+ T cells' metabolism is dysregulated in these pathologies and represents an opportunity for drug discovery and development. Genome-scale metabolic modeling offers an opportunity to accelerate drug discovery by providing high-quality information about possible target space in the context of a modeled disease. Here, we develop genome-scale models of naïve, Th1, Th2, and Th17 CD4+ T-cell subtypes to map metabolic perturbations in rheumatoid arthritis, multiple sclerosis, and primary biliary cholangitis. We subjected these models to in silico simulations for drug response analysis of existing FDA-approved drugs and compounds. Integration of disease-specific differentially expressed genes with altered reactions in response to metabolic perturbations identified 68 drug targets for the three autoimmune diseases. In vitro experimental validation, together with literature-based evidence, showed that modulation of fifty percent of identified drug targets suppressed CD4+ T cells, further increasing their potential impact as therapeutic interventions. Our approach can be generalized in the context of other diseases, and the metabolic models can be further used to dissect CD4+ T-cell metabolism.
Conflict of interest statement
The authors declare no competing interests.
Figures
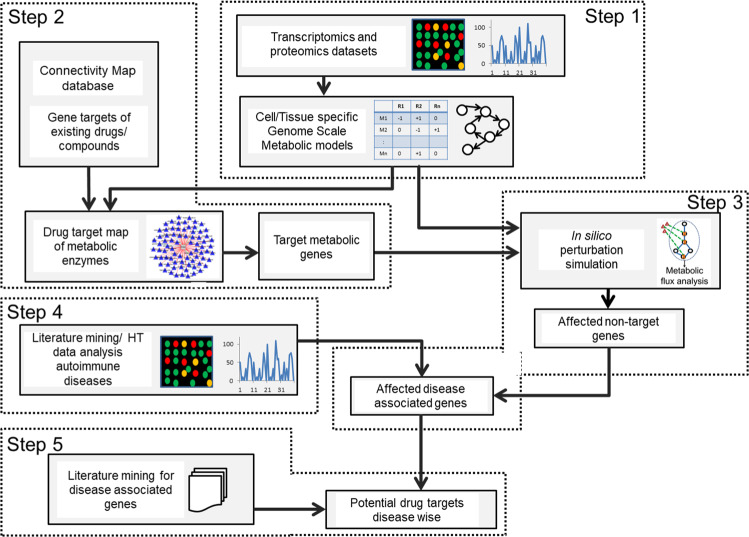
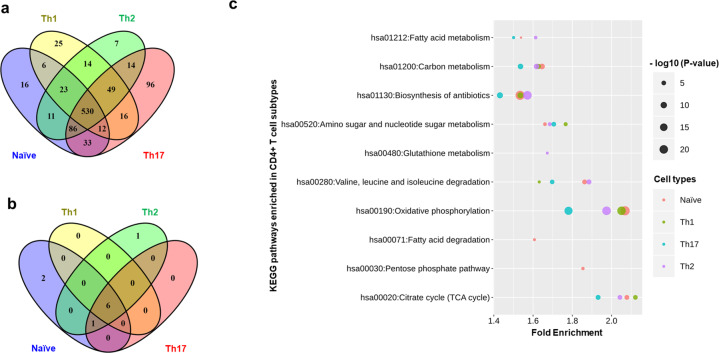
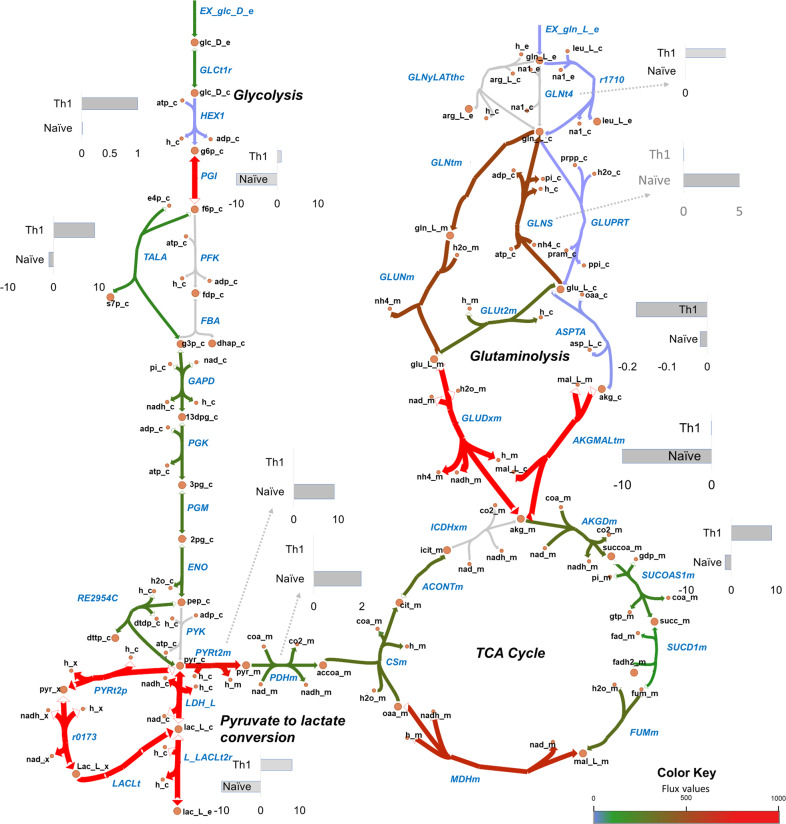
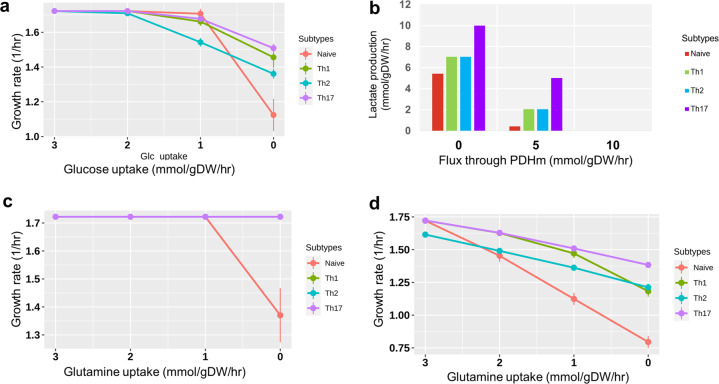
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
