

RUNX1/RUNX1T1 mediates alternative splicing and reorganises the transcriptional landscape in leukemia
- PMID: 33483506
- PMCID: PMC7822815
- DOI: 10.1038/s41467-020-20848-z
RUNX1/RUNX1T1 mediates alternative splicing and reorganises the transcriptional landscape in leukemia
Abstract
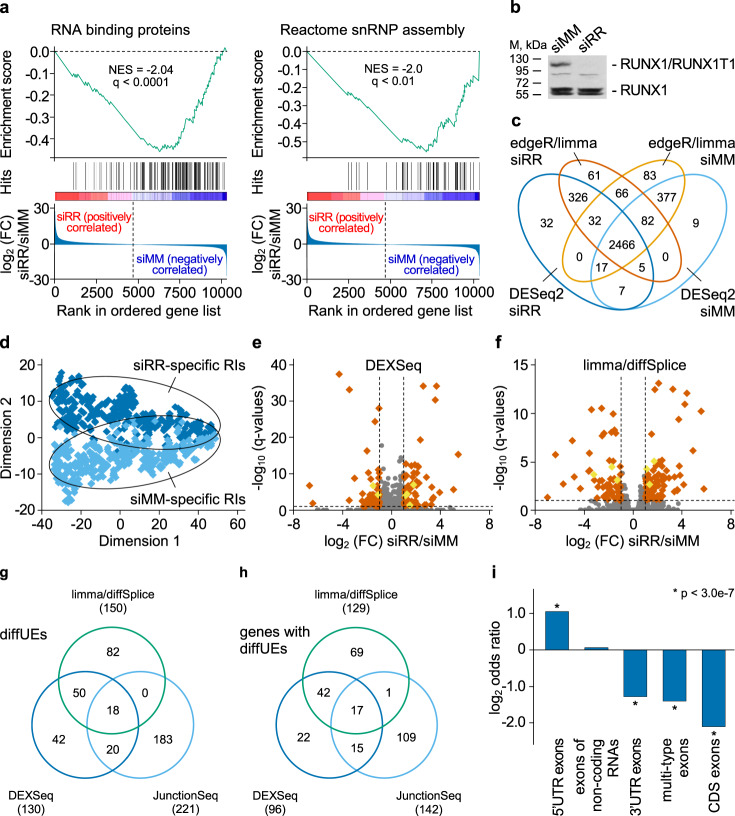
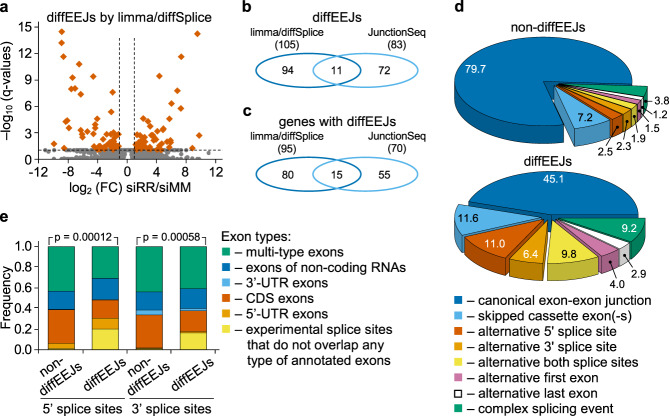
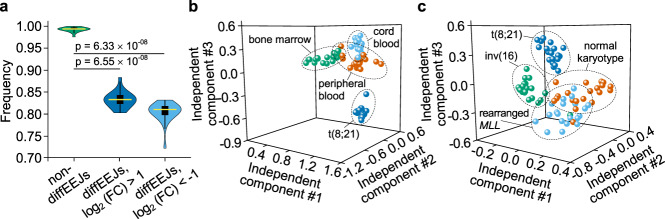
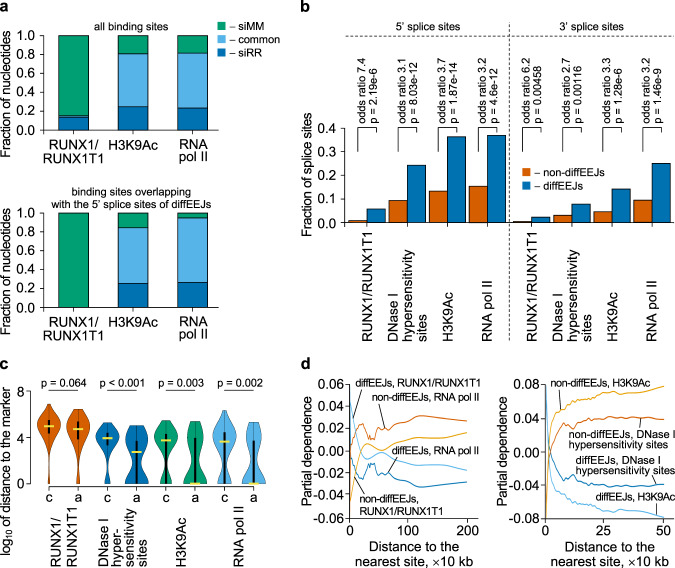
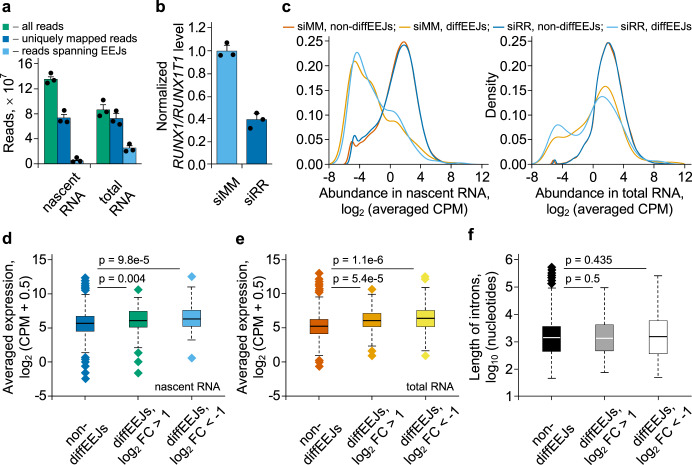
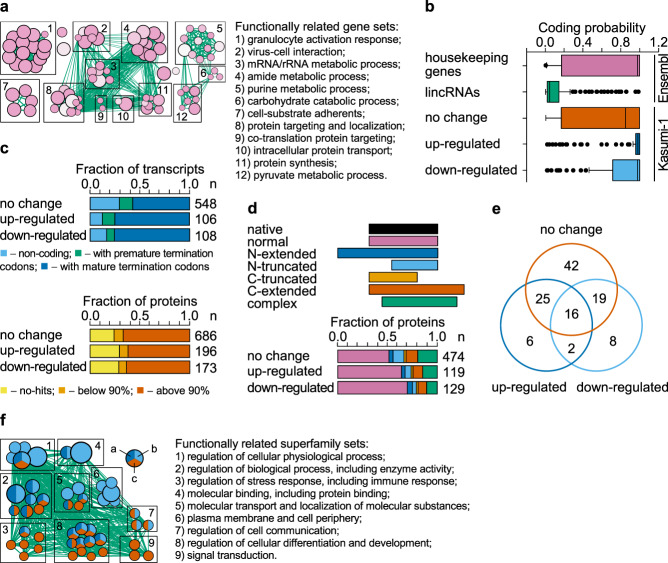
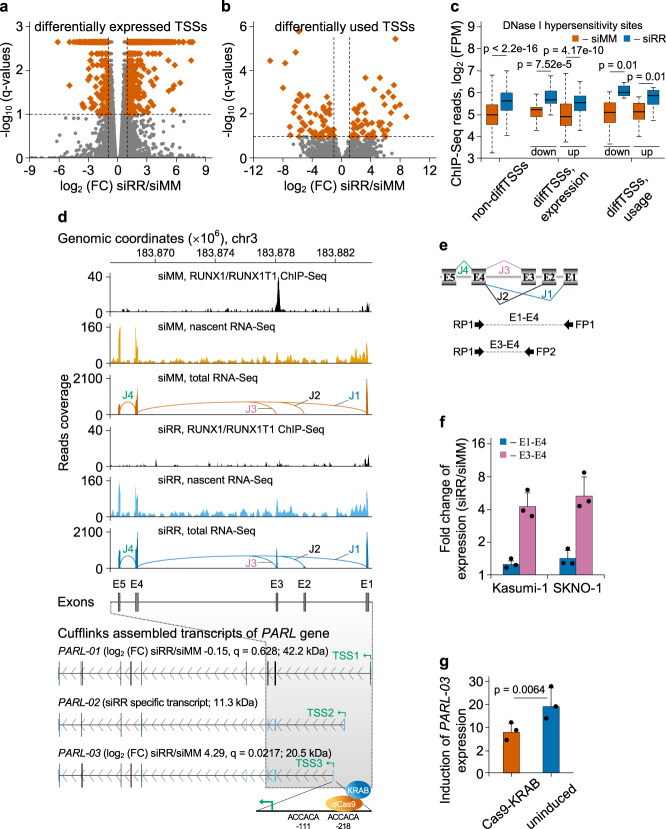
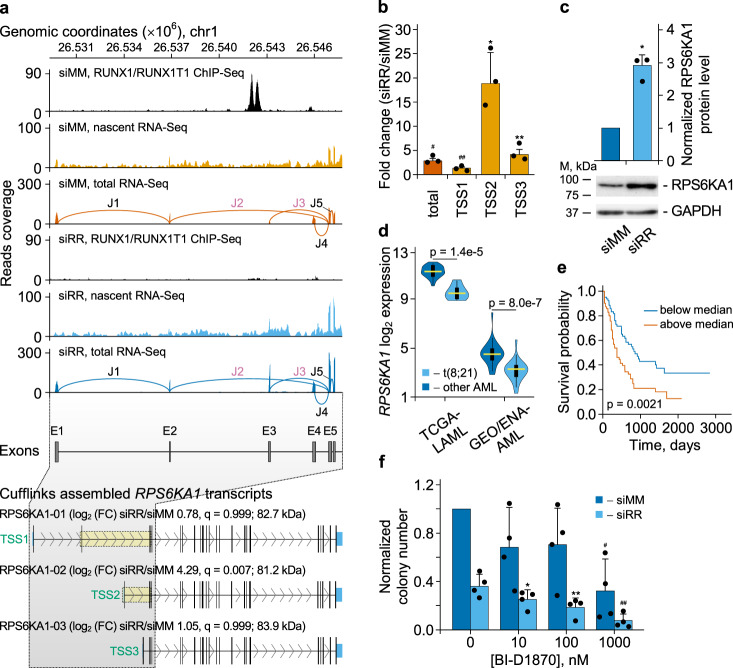
The fusion oncogene RUNX1/RUNX1T1 encodes an aberrant transcription factor, which plays a key role in the initiation and maintenance of acute myeloid leukemia. Here we show that the RUNX1/RUNX1T1 oncogene is a regulator of alternative RNA splicing in leukemic cells. The comprehensive analysis of RUNX1/RUNX1T1-associated splicing events identifies two principal mechanisms that underlie the differential production of RNA isoforms: (i) RUNX1/RUNX1T1-mediated regulation of alternative transcription start site selection, and (ii) direct or indirect control of the expression of genes encoding splicing factors. The first mechanism leads to the expression of RNA isoforms with alternative structure of the 5'-UTR regions. The second mechanism generates alternative transcripts with new junctions between internal cassettes and constitutive exons. We also show that RUNX1/RUNX1T1-mediated differential splicing affects several functional groups of genes and produces proteins with unique conserved domain structures. In summary, this study reveals alternative splicing as an important component of transcriptome re-organization in leukemia by an aberrant transcriptional regulator.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Decoding of exon splicing patterns in the human RUNX1-RUNX1T1 fusion gene.Int J Biochem Cell Biol. 2015 Nov;68:48-58. doi: 10.1016/j.biocel.2015.08.017. Epub 2015 Aug 29. Int J Biochem Cell Biol. 2015. PMID: 26320575
-
The RUNX1/RUNX1T1 network: translating insights into therapeutic options.Exp Hematol. 2021 Feb;94:1-10. doi: 10.1016/j.exphem.2020.11.005. Epub 2020 Nov 17. Exp Hematol. 2021. PMID: 33217477 Free PMC article. Review.
-
An AML1-ETO/miR-29b-1 regulatory circuit modulates phenotypic properties of acute myeloid leukemia cells.Oncotarget. 2017 Jun 20;8(25):39994-40005. doi: 10.18632/oncotarget.18127. Oncotarget. 2017. PMID: 28611288 Free PMC article.
-
Elucidation of Novel Therapeutic Targets for Acute Myeloid Leukemias with RUNX1-RUNX1T1 Fusion.Int J Mol Sci. 2019 Apr 6;20(7):1717. doi: 10.3390/ijms20071717. Int J Mol Sci. 2019. PMID: 30959925 Free PMC article.
-
RUNX1T1 function in cell fate.Stem Cell Res Ther. 2022 Jul 28;13(1):369. doi: 10.1186/s13287-022-03074-w. Stem Cell Res Ther. 2022. PMID: 35902872 Free PMC article. Review.
Cited by
-
RUNX1 deficiency cooperates with SRSF2 mutation to induce multilineage hematopoietic defects characteristic of MDS.Blood Adv. 2022 Dec 13;6(23):6078-6092. doi: 10.1182/bloodadvances.2022007804. Blood Adv. 2022. PMID: 36206200 Free PMC article.
-
Transcription Factors, R-Loops and Deubiquitinating Enzymes: Emerging Targets in Myelodysplastic Syndromes and Acute Myeloid Leukemia.Cancers (Basel). 2021 Jul 26;13(15):3753. doi: 10.3390/cancers13153753. Cancers (Basel). 2021. PMID: 34359655 Free PMC article. Review.
-
Nanoparticle-mediated targeting of the fusion gene RUNX1/ETO in t(8;21)-positive acute myeloid leukaemia.Leukemia. 2023 Apr;37(4):820-834. doi: 10.1038/s41375-023-01854-8. Epub 2023 Feb 23. Leukemia. 2023. PMID: 36823395 Free PMC article.
-
Identification of novel genes regulating the development of the palate.bioRxiv [Preprint]. 2024 Sep 15:2024.02.09.579685. doi: 10.1101/2024.02.09.579685. bioRxiv. 2024. Update in: Dev Dyn. 2025 Aug 2. doi: 10.1002/dvdy.70066. PMID: 38405938 Free PMC article. Updated. Preprint.
-
Detection and targeting of splicing deregulation in pediatric acute myeloid leukemia stem cells.Cell Rep Med. 2023 Mar 21;4(3):100962. doi: 10.1016/j.xcrm.2023.100962. Epub 2023 Mar 7. Cell Rep Med. 2023. PMID: 36889320 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
