

Family History of Pulmonary Fibrosis Predicts Worse Survival in Patients With Interstitial Lung Disease
- PMID: 33484728
- PMCID: PMC8173755
- DOI: 10.1016/j.chest.2021.01.026
Family History of Pulmonary Fibrosis Predicts Worse Survival in Patients With Interstitial Lung Disease
Abstract
Background: A number of genetic markers linked to familial pulmonary fibrosis predict differential survival in interstitial lung disease (ILD) patients. Although genetic testing is not performed routinely for ILD, family history commonly is obtained and may inform outcome risk.
Research question: Does survival vary between patients with and without self-reported familial pulmonary fibrosis?
Methods: Family history was acquired systematically for consecutive ILD patients who consented to clinical registry enrollment at the University of Texas Southwestern and the University of California at Davis. Patients were stratified by idiopathic pulmonary fibrosis (IPF) and non-IPF ILD diagnosis and were substratified by presence or absence of familial pulmonary fibrosis, defined as one or more additional affected family members. Transplant-free survival was compared using multilevel, mixed-effects Cox proportional hazards regression.
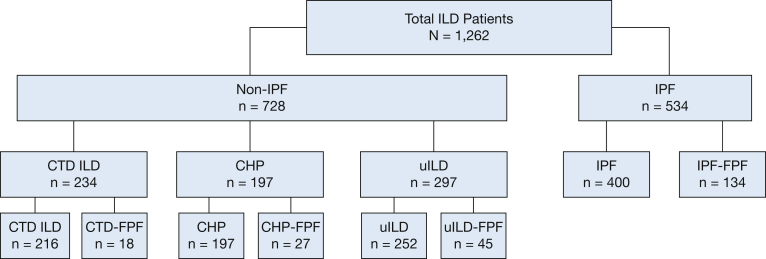
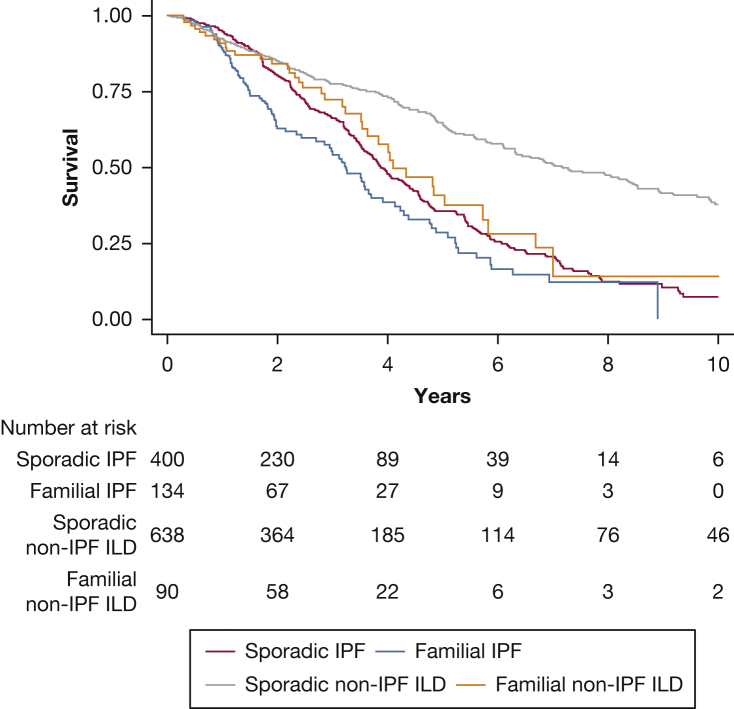
Results: Of the 1,262 patients included, 534 (42%) had IPF ILD and 728 (58%) had non-IPF ILD. Of those with non-IPF ILD, 18.5% had connective tissue disease, 15.6% had chronic hypersensitivity pneumonitis, and 23.5% had unclassifiable ILD. Familial pulmonary fibrosis was reported in 134 IPF ILD patients (25.1%) and 90 non-IPF ILD patients (12.4%). Those with familial IPF showed an 80% increased risk of death or transplantation compared with those with sporadic IPF (hazard ratio [HR], 1.8; 95% CI, 1.37-2.37; P < .001), whereas those with familial non-IPF ILD showed a twofold increased risk compared with their counterparts with sporadic disease (HR, 2.08; 95% CI, 1.46-2.96; P < .001). Outcome risk among those with familial non-IPF ILD was no different than for those with sporadic IPF ILD (HR, 1.27; 95% CI, 0.89-1.84; P = .19).
Interpretation: Patient-reported familial pulmonary fibrosis is predictive of reduced transplant-free survival in IPF and non-IPF ILD patients. Because survival among patients with familial non-IPF ILD approximates that of sporadic IPF ILD, early intervention should be considered for such patients. Until clinical genetic testing is widely available and provides actionable results, family history should be ascertained and considered in risk stratification.
Keywords: ILD; autoimmune; family history; hypersensitivity pneumonitis; pulmonary fibrosis; respiratory failure.
Copyright © 2021 American College of Chest Physicians. Published by Elsevier Inc. All rights reserved.
Figures
Comment in
-
A Comprehensive Family Health History and Genetic Analyses Are Complementary in the Detection of Risk for Progressive Pulmonary Fibrosis.Chest. 2021 May;159(5):1709-1710. doi: 10.1016/j.chest.2021.02.027. Chest. 2021. PMID: 33965125 No abstract available.
References
-
- Fischer A., du Bois R. Interstitial lung disease in connective tissue disorders. Lancet. 2012;380(9842):689–698. - PubMed
-
- Selman M., Pardo A., King T.E., Jr. Hypersensitivity pneumonitis: insights in diagnosis and pathobiology. Am J Respir Crit Care Med. 2012;186(4):314–324. - PubMed
-
- Lederer D.J., Martinez F.J. Idiopathic pulmonary fibrosis. N Engl J Med. 2018;378(19):1811–1823. - PubMed
-
- Neurohr C., Behr J. Changes in the current classification of IIP: a critical review. Respirology. 2015;20(5):699–704. - PubMed
-
- Bjoraker J.A., Ryu J.H., Edwin M.K. Prognostic significance of histopathologic subsets in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1998;157(1):199–203. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
