

An "Outside-In" and "Inside-Out" Consideration of Complement in the Multiple Sclerosis Brain: Lessons From Development and Neurodegenerative Diseases
- PMID: 33488361
- PMCID: PMC7817777
- DOI: 10.3389/fncel.2020.600656
An "Outside-In" and "Inside-Out" Consideration of Complement in the Multiple Sclerosis Brain: Lessons From Development and Neurodegenerative Diseases
Abstract
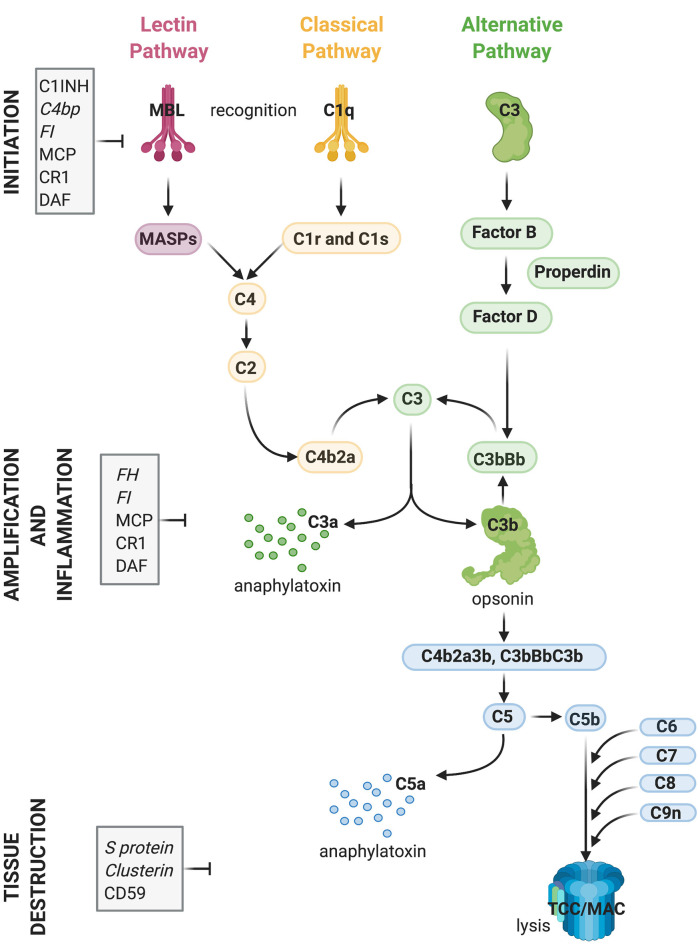
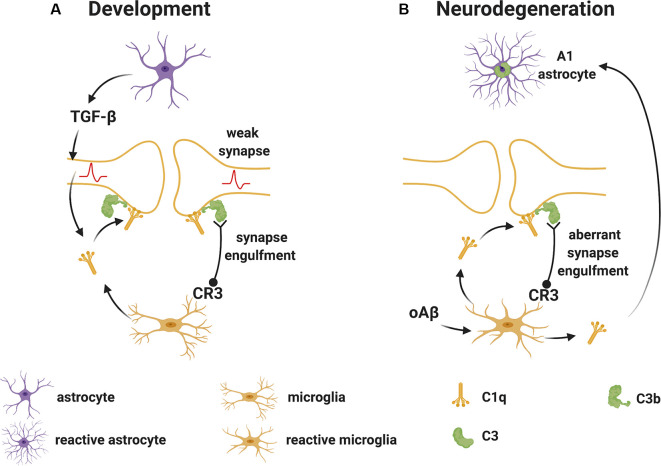
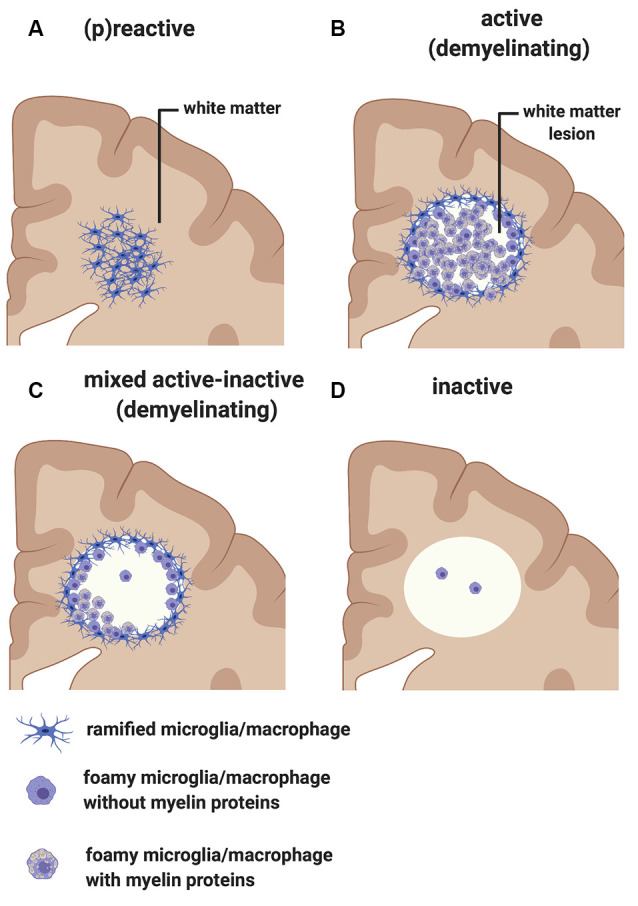
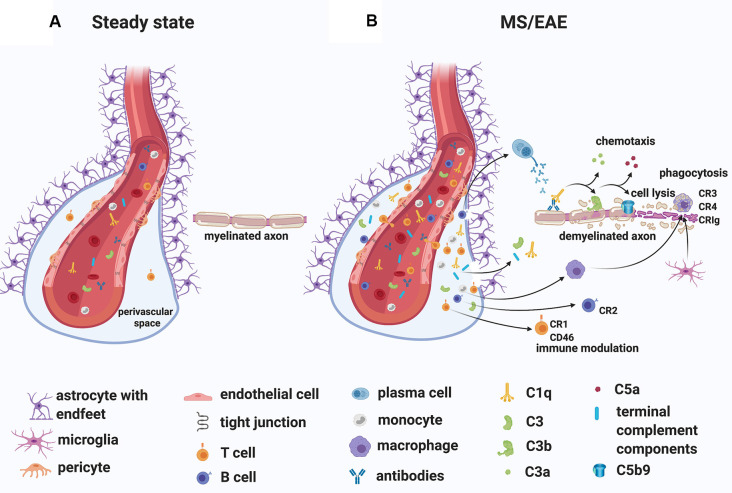
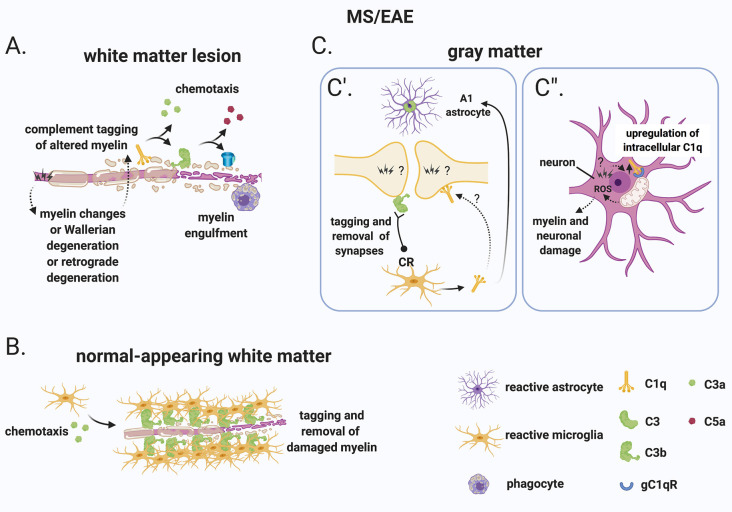
The last 15 years have seen an explosion of new findings on the role of complement, a major arm of the immune system, in the central nervous system (CNS) compartment including contributions to cell migration, elimination of synapse during development, aberrant synapse pruning in neurologic disorders, damage to nerve cells in autoimmune diseases, and traumatic injury. Activation of the complement system in multiple sclerosis (MS) is typically thought to occur as part of a primary (auto)immune response from the periphery (the outside) against CNS antigens (the inside). However, evidence of local complement production from CNS-resident cells, intracellular complement functions, and the more recently discovered role of early complement components in shaping synaptic circuits in the absence of inflammation opens up the possibility that complement-related sequelae may start and finish within the brain itself. In this review, the complement system will be introduced, followed by evidence that implicates complement in shaping the developing, adult, and normal aging CNS as well as its contribution to pathology in neurodegenerative conditions. Discussion of data supporting "outside-in" vs. "inside-out" roles of complement in MS will be presented, concluded by thoughts on potential approaches to therapies targeting specific elements of the complement system.
Keywords: complement; inside-out; multiple sclerosis; outside-in; pathology.
Copyright © 2021 Morgan, Gommerman and Ramaglia.
Conflict of interest statement
VR is co-inventor of a patent that describes the use of inhibitors of the terminal complement pathway for therapeutic purposes; she is co-founders of Regenesance B.V. and received consulting honorarium from EMD Serono and Fluidigm. JLG has received funding from Roche, Novartis and EMD Serono for research and funding from Roche for consulting activities. BPM is a consultant for RaPharma.
Figures
References
-
- Adams R. A., Bauer J., Flick M. J., Sikorski S. L., Nuriel T., Lassmann H., et al. (2007). The fibrin-derived γ377–395 peptide inhibits microglia activation and suppresses relapsing paralysis in central nervous system autoimmune disease. J. Exp. Med. 204, 571–582. 10.1084/jem.20061931 - DOI - PMC - PubMed
-
- Akassoglou K., Adams R. A., Bauer J., Mercado P., Tseveleki V., Lassmann H., et al. (2004). Fibrin depletion decreases inflammation and delays the onset of demyelination in a tumor necrosis factor transgenic mouse model for multiple sclerosis. Proc. Natl. Acad. Sci. U S A 101, 6698–6703. 10.1073/pnas.0303859101 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources