

Phosphonate and Bisphosphonate Inhibitors of Farnesyl Pyrophosphate Synthases: A Structure-Guided Perspective
- PMID: 33490038
- PMCID: PMC7815940
- DOI: 10.3389/fchem.2020.612728
Phosphonate and Bisphosphonate Inhibitors of Farnesyl Pyrophosphate Synthases: A Structure-Guided Perspective
Abstract
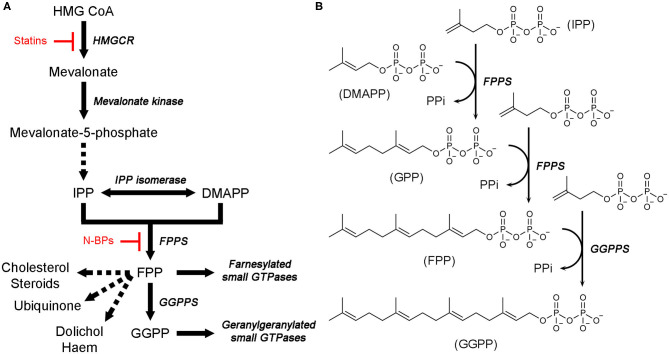
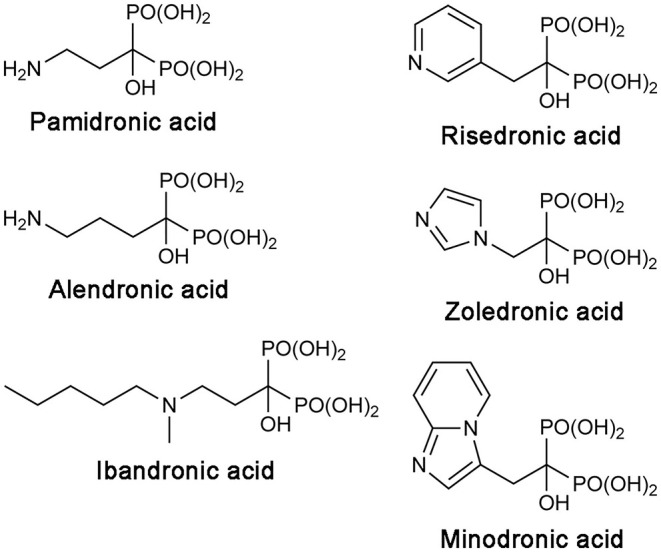
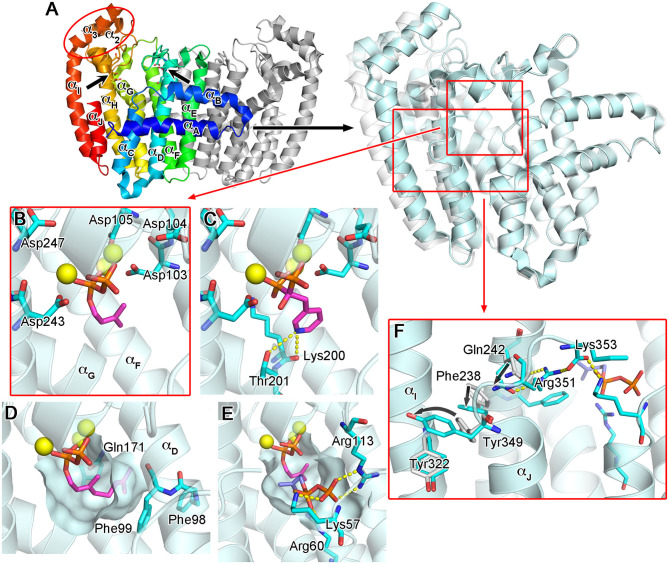
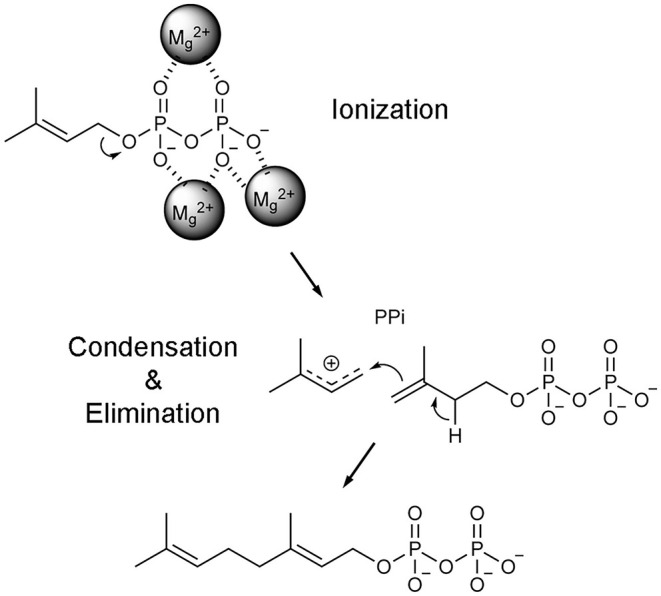
Phosphonates and bisphosphonates have proven their pharmacological utility as inhibitors of enzymes that metabolize phosphate and pyrophosphate substrates. The blockbuster class of drugs nitrogen-containing bisphosphonates represent one of the best-known examples. Widely used to treat bone-resorption disorders, these drugs work by inhibiting the enzyme farnesyl pyrophosphate synthase. Playing a key role in the isoprenoid biosynthetic pathway, this enzyme is also a potential anticancer target. Here, we provide a comprehensive overview of the research efforts to identify new inhibitors of farnesyl pyrophosphate synthase for various therapeutic applications. While the majority of these efforts have been directed against the human enzyme, some have been targeted on its homologs from other organisms, such as protozoan parasites and insects. Our particular focus is on the structures of the target enzymes and how the structural information has guided the drug discovery efforts.
Keywords: bisphosphonate; farnesyl pyrophosphate synthase; isoprenoid biosynthesis; phosphonate; structure-based drug design.
Copyright © 2021 Park, Pandya, Ezekiel and Berghuis.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
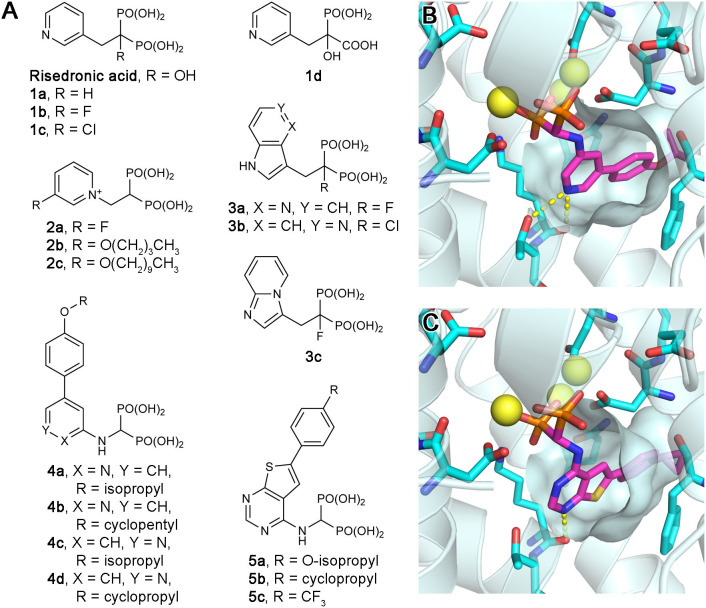
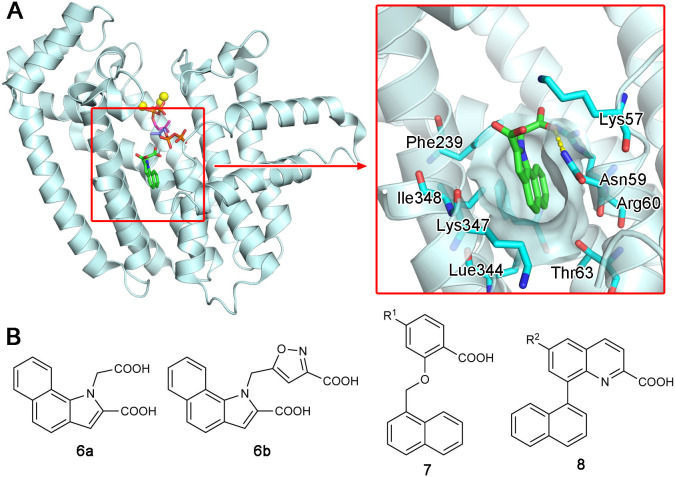
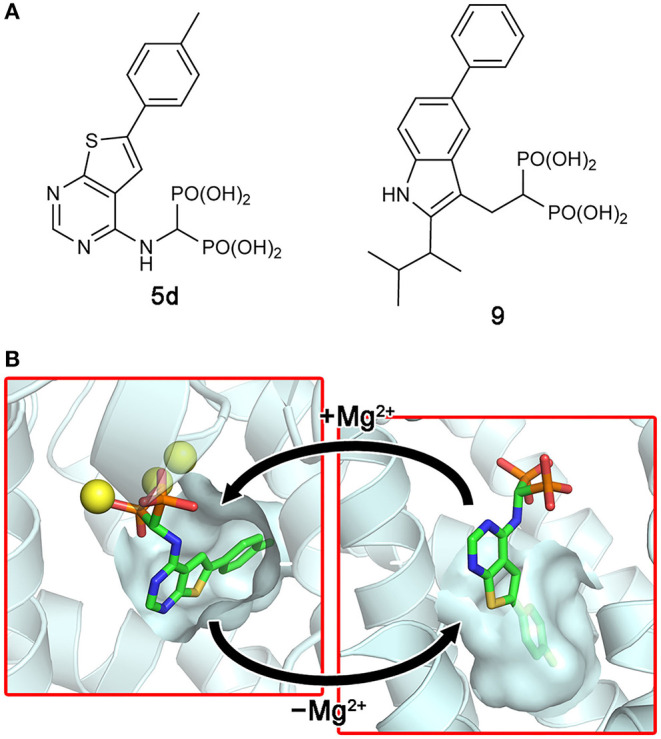
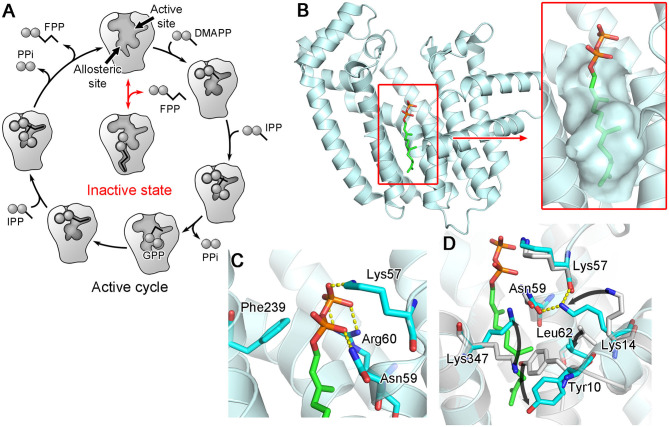
Similar articles
-
Approaches for Designing new Potent Inhibitors of Farnesyl Pyrophosphate Synthase.Expert Opin Drug Discov. 2016;11(3):307-20. doi: 10.1517/17460441.2016.1143814. Epub 2016 Feb 15. Expert Opin Drug Discov. 2016. PMID: 26781029 Review.
-
Structural characterization of substrate and inhibitor binding to farnesyl pyrophosphate synthase from Pseudomonas aeruginosa.Acta Crystallogr D Biol Crystallogr. 2015 Mar;71(Pt 3):721-31. doi: 10.1107/S1399004715001121. Epub 2015 Feb 26. Acta Crystallogr D Biol Crystallogr. 2015. PMID: 25760619 Free PMC article.
-
Molecular characterization of farnesyl pyrophosphate synthase from Bacopa monniera by comparative modeling and docking studies.Bioinformation. 2012;8(22):1075-81. doi: 10.6026/97320630081075. Epub 2012 Nov 13. Bioinformation. 2012. PMID: 23251041 Free PMC article.
-
Structural basis for bisphosphonate-mediated inhibition of isoprenoid biosynthesis.J Biol Chem. 2004 Mar 5;279(10):8526-9. doi: 10.1074/jbc.C300511200. Epub 2003 Dec 12. J Biol Chem. 2004. PMID: 14672944
-
The relationship between the chemistry and biological activity of the bisphosphonates.Bone. 2011 Jul;49(1):20-33. doi: 10.1016/j.bone.2011.03.774. Epub 2011 Apr 9. Bone. 2011. PMID: 21497677 Review.
Cited by
-
Bisphosphonates: The role of chemistry in understanding their biological actions and structure-activity relationships, and new directions for their therapeutic use.Bone. 2022 Mar;156:116289. doi: 10.1016/j.bone.2021.116289. Epub 2021 Dec 8. Bone. 2022. PMID: 34896359 Free PMC article.
-
How zoledronic acid improves osteoporosis by acting on osteoclasts.Front Pharmacol. 2022 Aug 25;13:961941. doi: 10.3389/fphar.2022.961941. eCollection 2022. Front Pharmacol. 2022. PMID: 36091799 Free PMC article. Review.
-
Impact of Vitamin D deficiency on immunological and metabolic responses in women with recurrent pregnancy loss: focus on VDBP/HLA-G1/CTLA-4/ENTPD1/adenosine-fetal-maternal conflict crosstalk.BMC Pregnancy Childbirth. 2024 Oct 29;24(1):709. doi: 10.1186/s12884-024-06914-0. BMC Pregnancy Childbirth. 2024. PMID: 39472874 Free PMC article.
-
Hydroxy- and Amino-Phosphonates and -Bisphosphonates: Synthetic Methods and Their Biological Applications.Front Chem. 2022 Jun 1;10:890696. doi: 10.3389/fchem.2022.890696. eCollection 2022. Front Chem. 2022. PMID: 35721002 Free PMC article. Review.
-
A Systematic Review on the Efficacy of Bisphosphonates on Osteogenesis Imperfecta.Cureus. 2025 Jun 22;17(6):e86549. doi: 10.7759/cureus.86549. eCollection 2025 Jun. Cureus. 2025. PMID: 40698241 Free PMC article. Review.
References
-
- Amstutz R., Bold G., Cotesta S., Jahnke W., Marzinzik A., Hartwieg J. C. D., et al. . (2012). Quinolines as Inhibitors of Farnesyl Pyrophosphate Synthase. Google Patents. - PubMed
-
- Aripirala S., Gonzalez-Pacanowska D., Oldfield E., Kaiser M., Amzel L. M., Gabelli S. B. (2014). Structural and thermodynamic basis of the inhibition of Leishmania major farnesyl diphosphate synthase by nitrogen-containing bisphosphonates. Acta Crystallogr. D Biol. Crystallogr. 70, 802–810. 10.1107/S1399004713033221 - DOI - PMC - PubMed
-
- Aripirala S., Szajnman S. H., Jakoncic J., Rodriguez J. B., Docampo R., Gabelli S. B., et al. . (2012). Design, synthesis, calorimetry, and crystallographic analysis of 2-alkylaminoethyl-1,1-bisphosphonates as inhibitors of Trypanosoma cruzi farnesyl diphosphate synthase. J. Med. Chem. 55, 6445–6454. 10.1021/jm300425y - DOI - PMC - PubMed
-
- Benford H. L., Frith J. C., Auriola S., Monkkonen J., Rogers M. J. (1999). Farnesol and geranylgeraniol prevent activation of caspases by aminobisphosphonates: biochemical evidence for two distinct pharmacological classes of bisphosphonate drugs. Mol. Pharmacol. 56, 131–140. 10.1124/mol.56.1.131 - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources