

Regional brain iron associated with deterioration in Alzheimer's disease: A large cohort study and theoretical significance
- PMID: 33491917
- PMCID: PMC9701539
- DOI: 10.1002/alz.12282
Regional brain iron associated with deterioration in Alzheimer's disease: A large cohort study and theoretical significance
Abstract
Objective: This paper is a proposal for an update of the iron hypothesis of Alzheimer's disease (AD), based on large-scale emerging evidence.
Background: Iron featured historically early in AD research efforts for its involvement in the amyloid and tau proteinopathies, APP processing, genetics, and one clinical trial, yet iron neurochemistry remains peripheral in mainstream AD research. Much of the effort investigating iron in AD has focused on the potential for iron to provoke the onset of disease, by promoting proteinopathy though increased protein expression, phosphorylation, and aggregation.
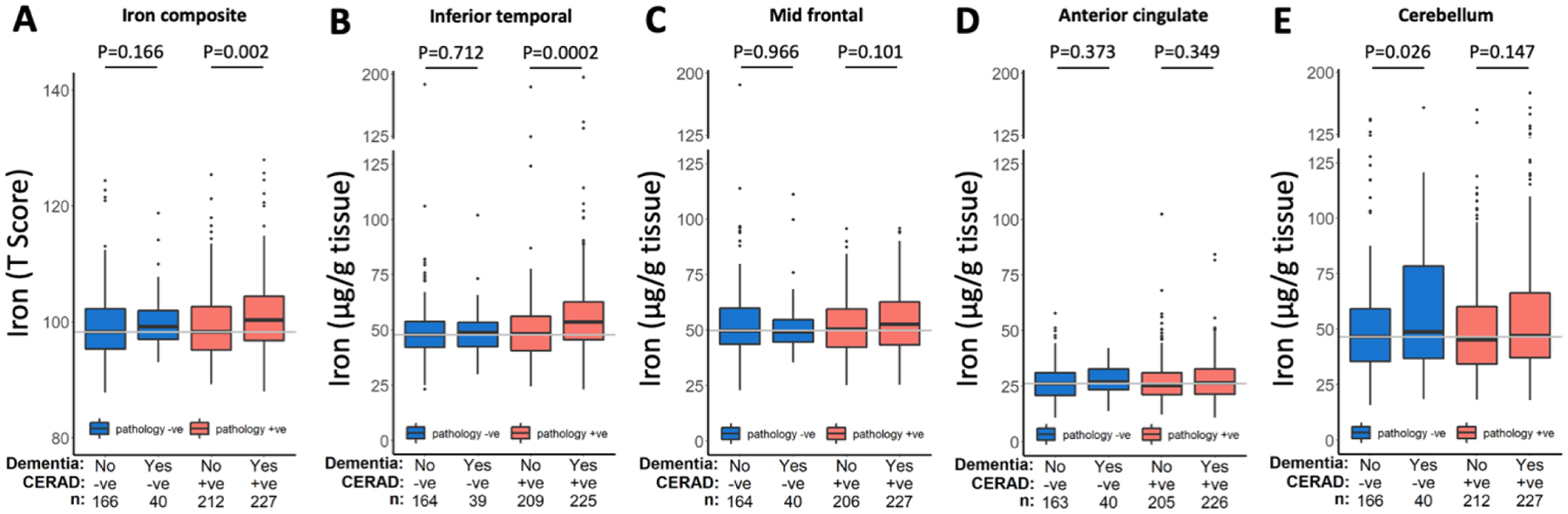
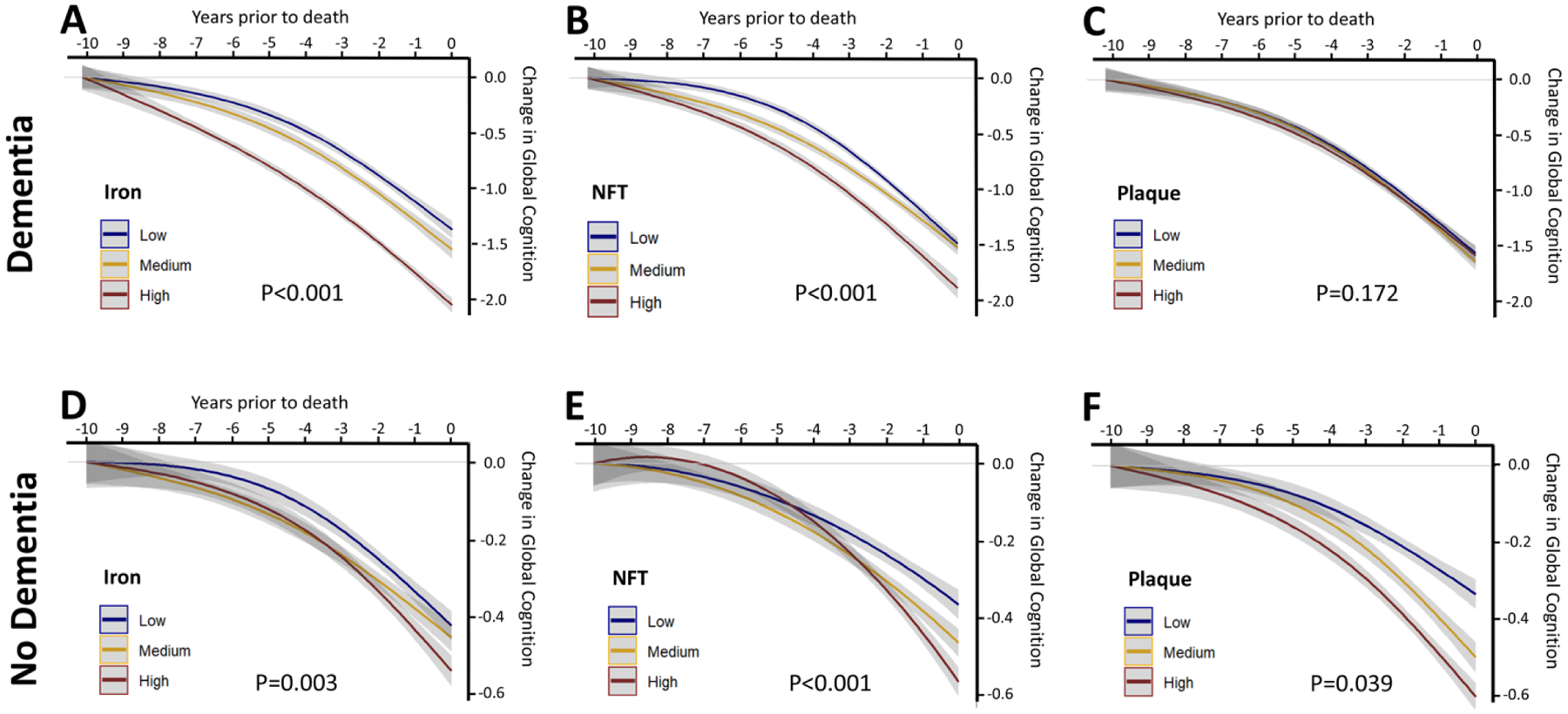
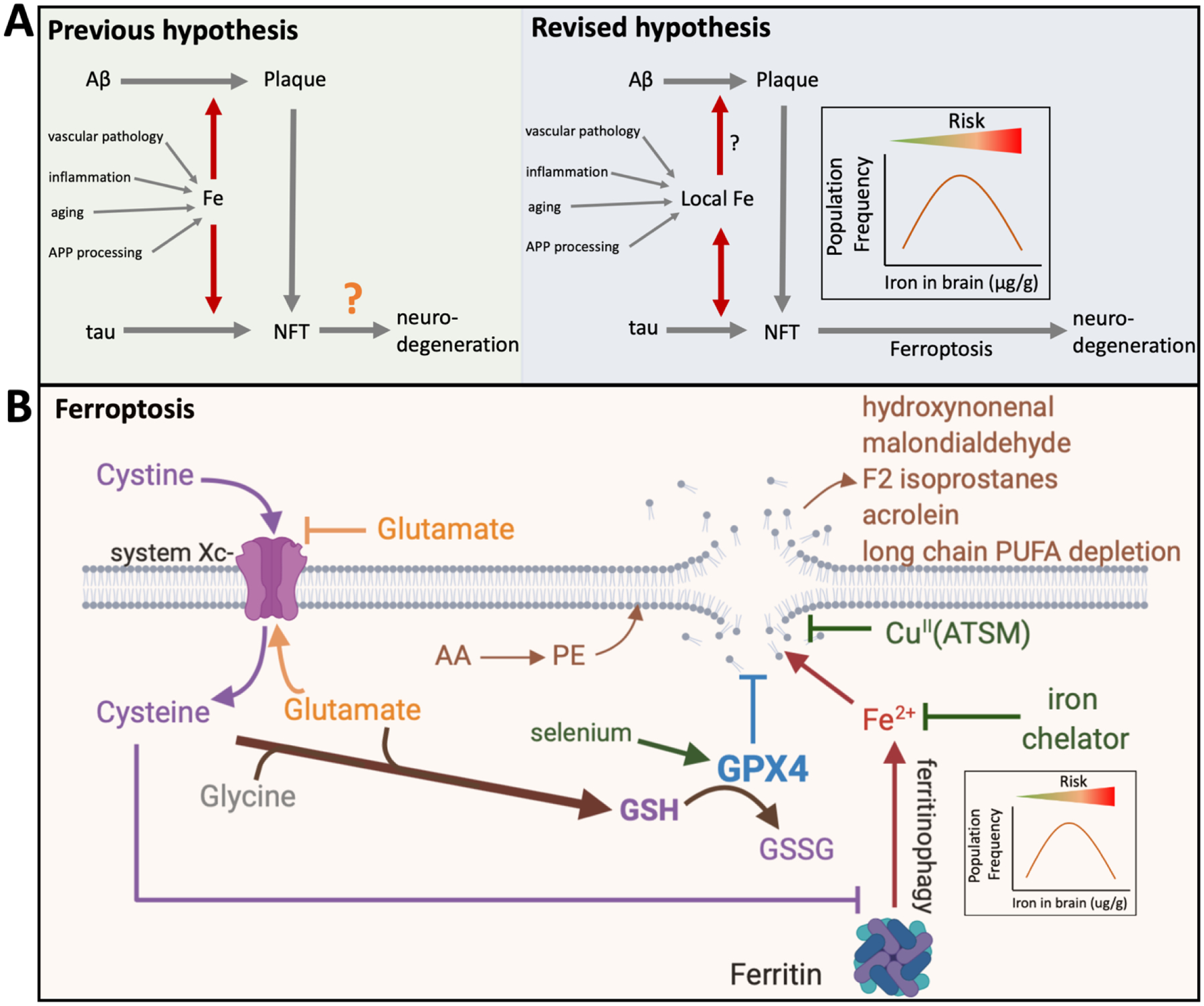
New/updated hypothesis: We provide new evidence from a large post mortem cohort that brain iron levels within the normal range were associated with accelerated ante mortem disease progression in cases with underlying proteinopathic neuropathology. These results corroborate recent findings that argue for an additional downstream role for iron as an effector of neurodegeneration, acting independently of tau or amyloid pathologies. We hypothesize that the level of tissue iron is a trait that dictates the probability of neurodegeneration in AD by ferroptosis, a regulated cell death pathway that is initiated by signals such as glutathione depletion and lipid peroxidation.
Major challenges for the hypothesis: While clinical biomarkers of ferroptosis are still in discovery, the demonstration of additional ferroptotic correlates (genetic or biomarker derived) of disease progression is required to test this hypothesis. The genes implicated in familial AD are not known to influence ferroptosis, although recent reports on APP mutations and apolipoprotein E allele (APOE) have shown impact on cellular iron retention. Familial AD mutations will need to be tested for their impact on ferroptotic vulnerability. Ultimately, this hypothesis will be substantiated, or otherwise, by a clinical trial of an anti-ferroptotic/iron compound in AD patients.
Linkage to other major theories: Iron has historically been linked to the amyloid and tau proteinopathies of AD. Tau, APP, and apoE have been implicated in physiological iron homeostasis in the brain. Iron is biochemically the origin of most chemical radicals generated in biochemistry and thus closely associated with the oxidative stress theory of AD. Iron accumulation is also a well-established consequence of aging and inflammation, which are major theories of disease pathogenesis.
Keywords: Alzheimer's disease; cognitive decline; iron; neurodegeneration; pathology.
© 2021 the Alzheimer's Association.
Conflict of interest statement
Conflict of interest statement
AIB is a shareholder in Prana Biotechnology Ltd, Cogstate Ltd, Brighton Biotech LLC, Grunbiotics Pty Ltd, Eucalyptus Pty Ltd, and Mesoblast Ltd. He is a paid consultant for, and has a profit share interest in, Collaborative Medicinal Development Pty Ltd.
Figures
References
-
- Goodman L. Alzheimer’s disease; a clinico-pathologic analysis of twenty-three cases with a theory on pathogenesis. The Journal of nervous and mental disease. 1953;118:97–130. - PubMed
-
- Hallgren B, Sourander P. The non-haemin iron in the cerebral cortex in Alzheimer’s disease. J Neurochem. 1960;5:307–10. - PubMed
-
- Crapper McLachlan DR, Dalton AJ, Kruck TP, Bell MY, Smith WL, Kalow W, et al. Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet. 1991;337:1304–8. - PubMed
-
- Connor JR, Menzies SL, St Martin SM, Mufson EJ. A histochemical study of iron, transferrin, and ferritin in Alzheimer’s diseased brains. Journal of neuroscience research. 1992;31:75–83. - PubMed
-
- Connor JR, Snyder BS, Beard JL, Fine RE, Mufson EJ. Regional distribution of iron and iron-regulatory proteins in the brain in aging and Alzheimer’s disease. Journal of neuroscience research. 1992;31:327–35. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
