

Caveolae-mediated Tie2 signaling contributes to CCM pathogenesis in a brain endothelial cell-specific Pdcd10-deficient mouse model
- PMID: 33495460
- PMCID: PMC7835246
- DOI: 10.1038/s41467-020-20774-0
Caveolae-mediated Tie2 signaling contributes to CCM pathogenesis in a brain endothelial cell-specific Pdcd10-deficient mouse model
Erratum in
-
Author Correction: Caveolae-mediated Tie2 signaling contributes to CCM pathogenesis in a brain endothelial cell-specific Pdcd10-deficient mouse model.Nat Commun. 2025 Jul 10;16(1):6352. doi: 10.1038/s41467-025-61617-0. Nat Commun. 2025. PMID: 40640167 Free PMC article. No abstract available.
Abstract
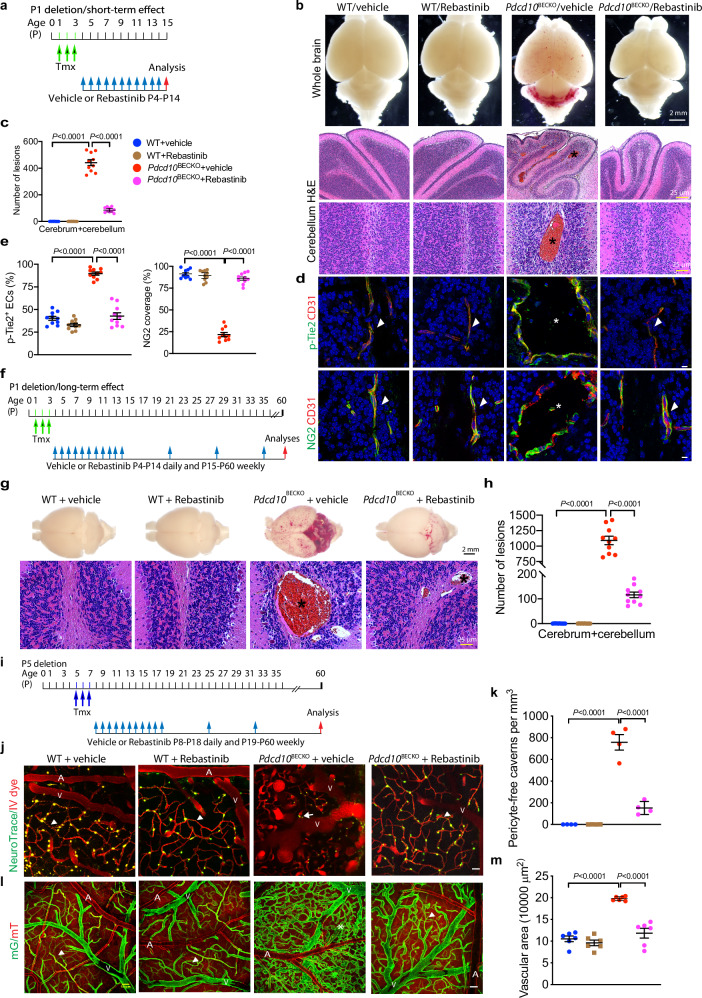
Cerebral cavernous malformations (CCMs) are vascular abnormalities that primarily occur in adulthood and cause cerebral hemorrhage, stroke, and seizures. CCMs are thought to be initiated by endothelial cell (EC) loss of any one of the three Ccm genes: CCM1 (KRIT1), CCM2 (OSM), or CCM3 (PDCD10). Here we report that mice with a brain EC-specific deletion of Pdcd10 (Pdcd10BECKO) survive up to 6-12 months and develop bona fide CCM lesions in all regions of brain, allowing us to visualize the vascular dynamics of CCM lesions using transcranial two-photon microscopy. This approach reveals that CCMs initiate from protrusion at the level of capillary and post-capillary venules with gradual dissociation of pericytes. Microvascular beds in lesions are hyper-permeable, and these disorganized structures present endomucin-positive ECs and α-smooth muscle actin-positive pericytes. Caveolae in the endothelium of Pdcd10BECKO lesions are drastically increased, enhancing Tie2 signaling in Ccm3-deficient ECs. Moreover, genetic deletion of caveolin-1 or pharmacological blockade of Tie2 signaling effectively normalizes microvascular structure and barrier function with attenuated EC-pericyte disassociation and CCM lesion formation in Pdcd10BECKO mice. Our study establishes a chronic CCM model and uncovers a mechanism by which CCM3 mutation-induced caveolae-Tie2 signaling contributes to CCM pathogenesis.
Conflict of interest statement
The authors declare no competing interests.
Figures









References
-
- Otten, P., Pizzolato, G. P., Rilliet, B. & Berney, J. 131 cases of cavernous angioma (cavernomas) of the CNS, discovered by retrospective analysis of 24,535 autopsies. Neurochirurgie35(82-83), 128–131 (1989). - PubMed
-
- Rigamonti, D. et al. Cerebral cavernous malformations. N. Engl. J. Med.319, 343–347 (1988). - PubMed
-
- Sahoo, T. et al. Mutations in the gene encoding KRIT1, a Krev-1/rap1a binding protein, cause cerebral cavernous malformations (CCM1). Hum. Mol. Genet.8, 2325–2333 (1999). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
