

Transforming the clinical outcome in CRIM-negative infantile Pompe disease identified via newborn screening: the benefits of early treatment with enzyme replacement therapy and immune tolerance induction
- PMID: 33495531
- PMCID: PMC8107133
- DOI: 10.1038/s41436-020-01080-y
Transforming the clinical outcome in CRIM-negative infantile Pompe disease identified via newborn screening: the benefits of early treatment with enzyme replacement therapy and immune tolerance induction
Abstract
Purpose: To assess the magnitude of benefit to early treatment initiation, enabled by newborn screening or prenatal diagnosis, in patients with cross-reactive immunological material (CRIM)-negative infantile Pompe disease (IPD), treated with enzyme replacement therapy (ERT) and prophylactic immune tolerance induction (ITI) with rituximab, methotrexate, and intravenous immunoglobulin (IVIG).
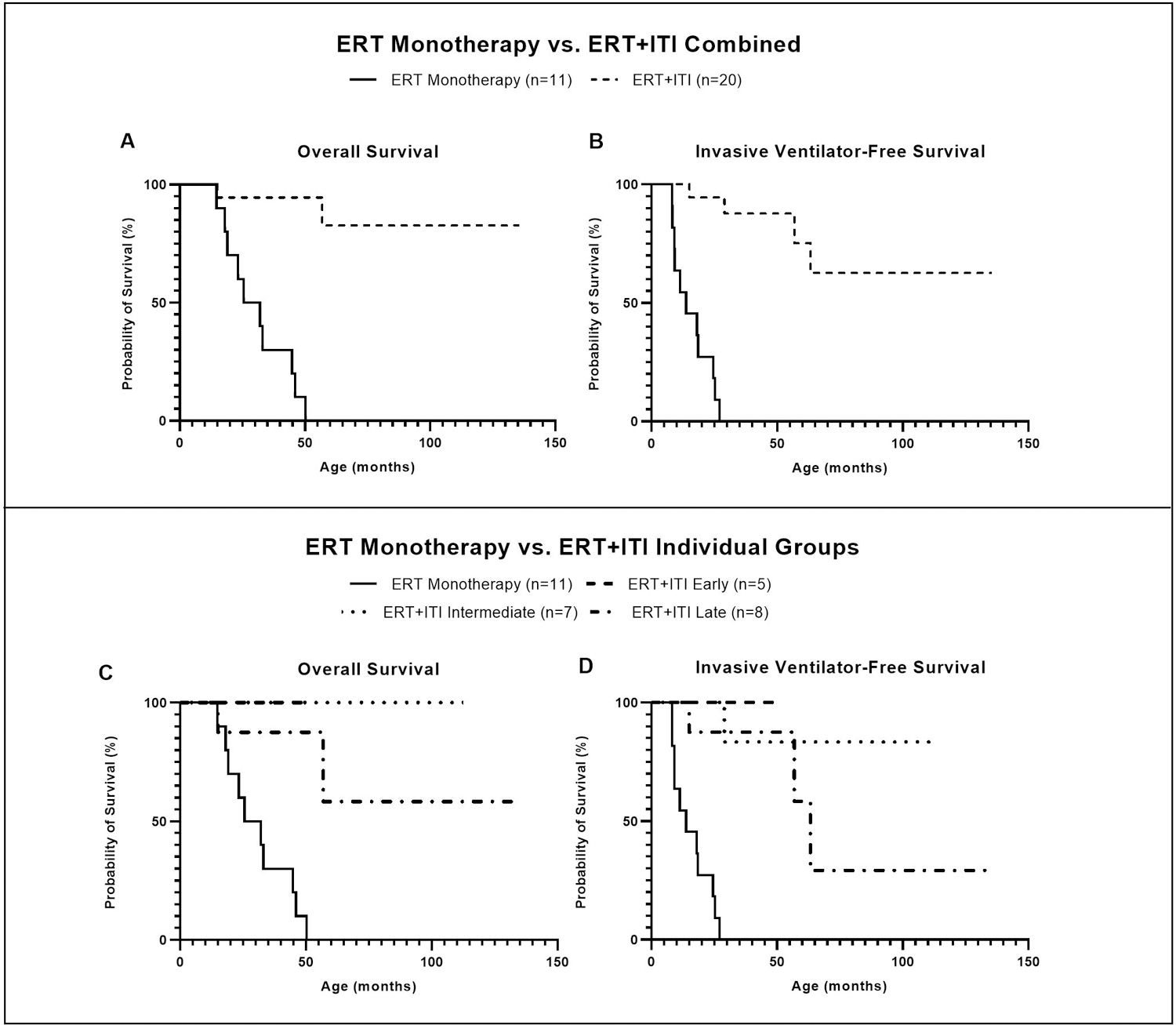
Methods: A total of 41 CRIM-negative IPD patients were evaluated. Among patients who were treated with ERT + ITI (n = 30), those who were invasive ventilator-free at baseline and had ≥6 months of follow-up were stratified based on age at treatment initiation: (1) early (≤4 weeks), (2) intermediate (>4 and ≤15 weeks), and (3) late (>15 weeks). A historical cohort of 11 CRIM-negative patients with IPD treated with ERT monotherapy served as an additional comparator group.
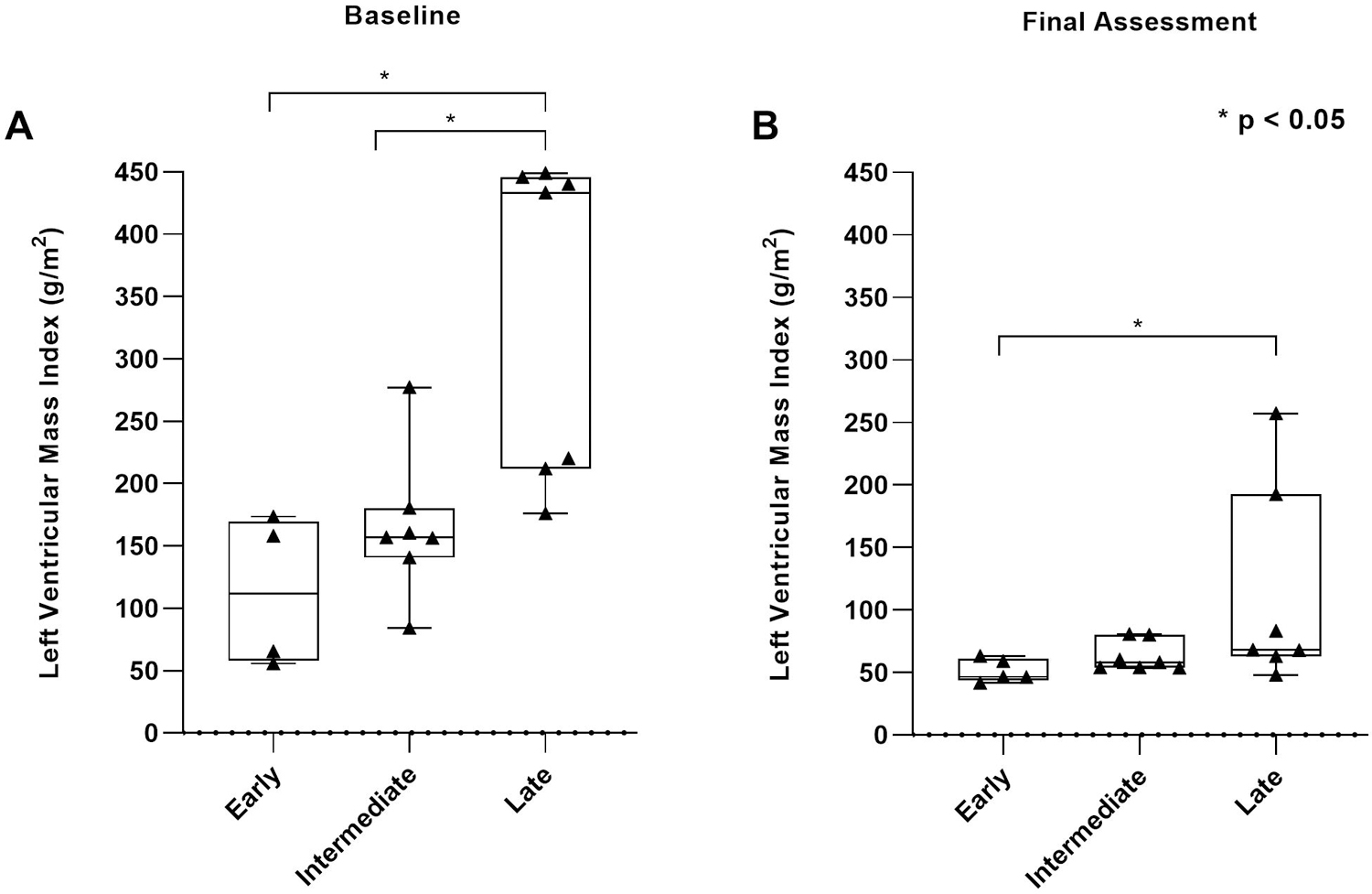
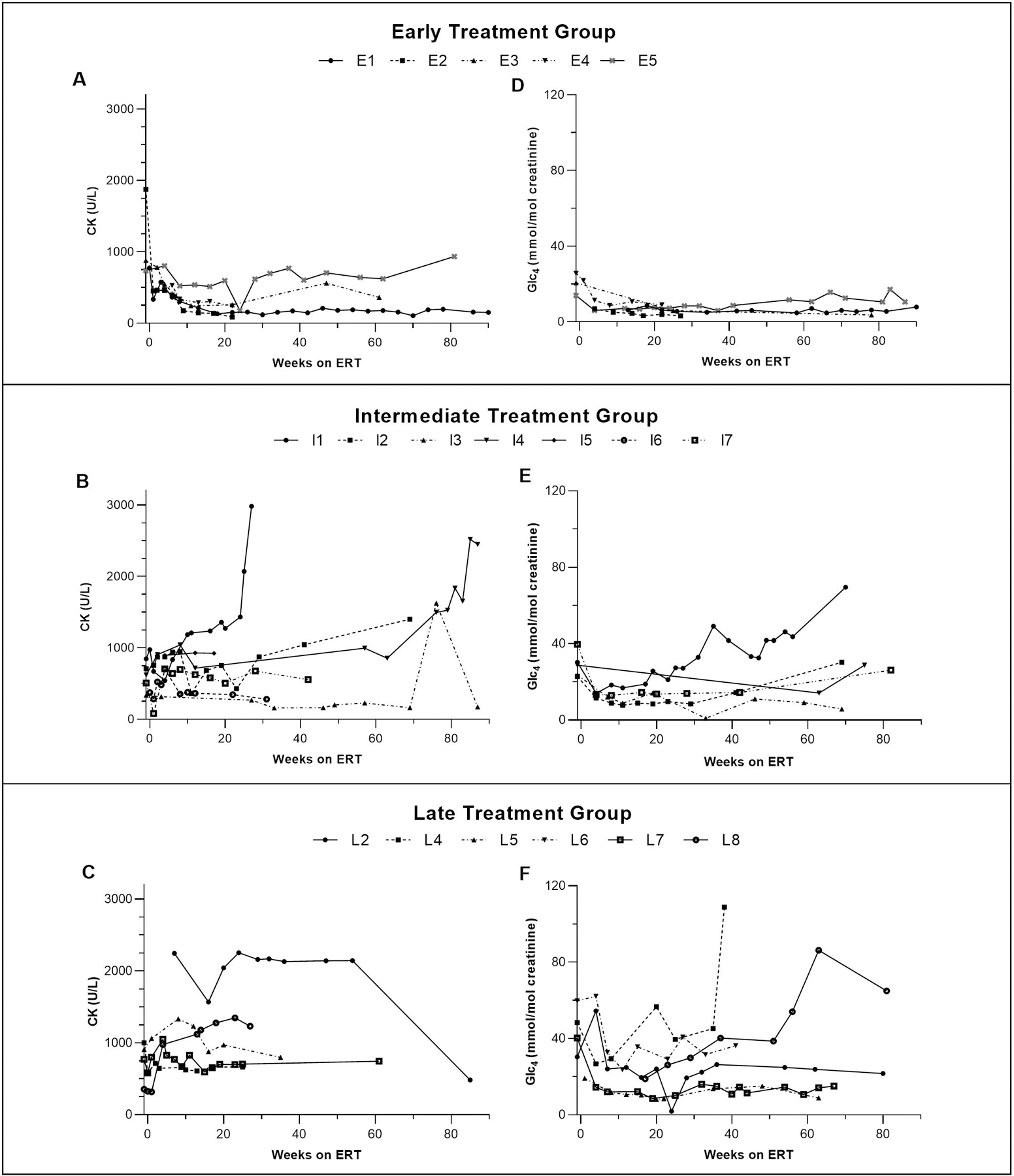
Results: Twenty patients were included; five, seven, and eight in early, intermediate, and late treatment groups, respectively. Genotypes were similar across the three groups. Early-treated patients showed significant improvements in left ventricular mass index, motor and pulmonary outcomes, as well as biomarkers creatine kinase and urinary glucose tetrasaccharide, compared with those treated later.
Conclusion: Our preliminary data suggest that early treatment with ERT + ITI can transform the long-term CRIM-negative IPD phenotype, which represents the most severe end of the Pompe disease spectrum.
Conflict of interest statement
CONFLICT OF INTEREST
Figures
References
-
- Hirschhorn R, Reuser A. Glycogen storage disease type II; acid alpha-glucosidase (acid maltase) deficiency. The Metabolic and Molecular Bases of Inherited Disease 8th edition. Edited by: Scriver CR, Beaudet AL, Sly WS, Valle D. 2001. In: McGraw-Hill, New York.
-
- van den Hout HM, Hop W, van Diggelen OP, et al. The natural course of infantile Pompe’s disease: 20 original cases compared with 133 cases from the literature. Pediatrics. 2003;112(2):332–340. - PubMed
-
- Kishnani PS, Hwu W-L, Mandel H, Nicolino M, Yong F, Corzo D. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. The Journal of Pediatrics. 2006;148(5):671–676.e672. - PubMed
-
- Amalfitano A, Bengur AR, Morse RP, et al. Recombinant human acid alpha-glucosidase enzyme therapy for infantile glycogen storage disease type II: results of a phase I/II clinical trial. Genet Med. 2001;3(2):132–138. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
