

Genetic Disease and Therapy
- PMID: 33497260
- PMCID: PMC9704033
- DOI: 10.1146/annurev-pathmechdis-012419-032626
Genetic Disease and Therapy
Abstract
Genetic diseases cause numerous complex and intractable pathologies. DNA sequences encoding each human's complexity and many disease risks are contained in the mitochondrial genome, nuclear genome, and microbial metagenome. Diagnosis of these diseases has unified around applications of next-generation DNA sequencing. However, translating specific genetic diagnoses into targeted genetic therapies remains a central goal. To date, genetic therapies have fallen into three broad categories: bulk replacement of affected genetic compartments with a new exogenous genome, nontargeted addition of exogenous genetic material to compensate for genetic errors, and most recently, direct correction of causative genetic alterations using gene editing. Generalized methods of diagnosis, therapy, and reagent delivery into each genetic compartment will accelerate the next generations of curative genetic therapies. We discuss the structure and variability of the mitochondrial, nuclear, and microbial metagenomic compartments, as well as the historical development and current practice of genetic diagnostics and gene therapies targeting each compartment.
Keywords: clinical genetics; gene editing; gene therapies; genetic diagnostics; genetic disease.
Figures
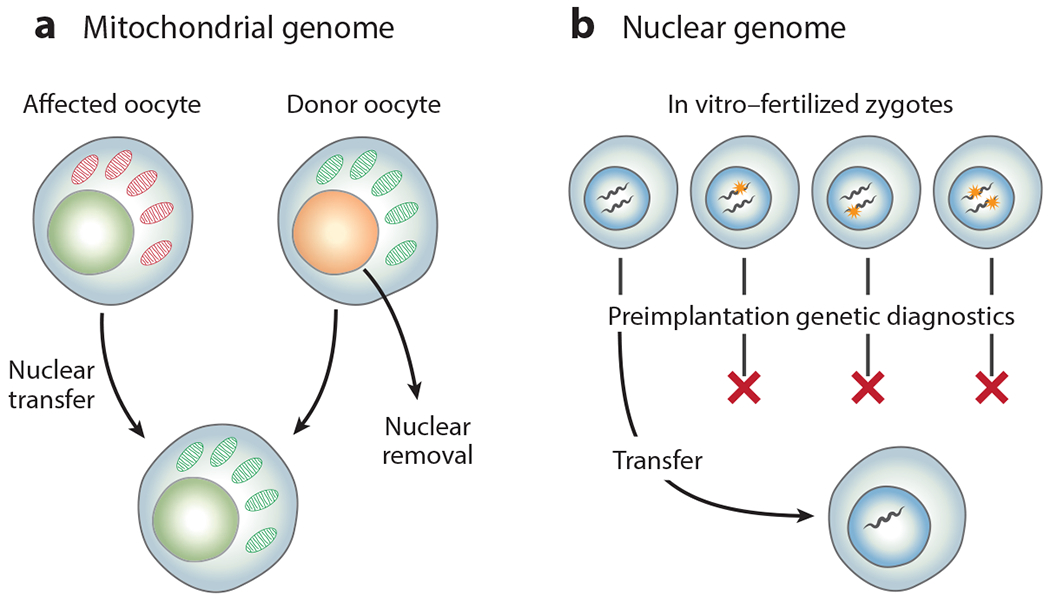
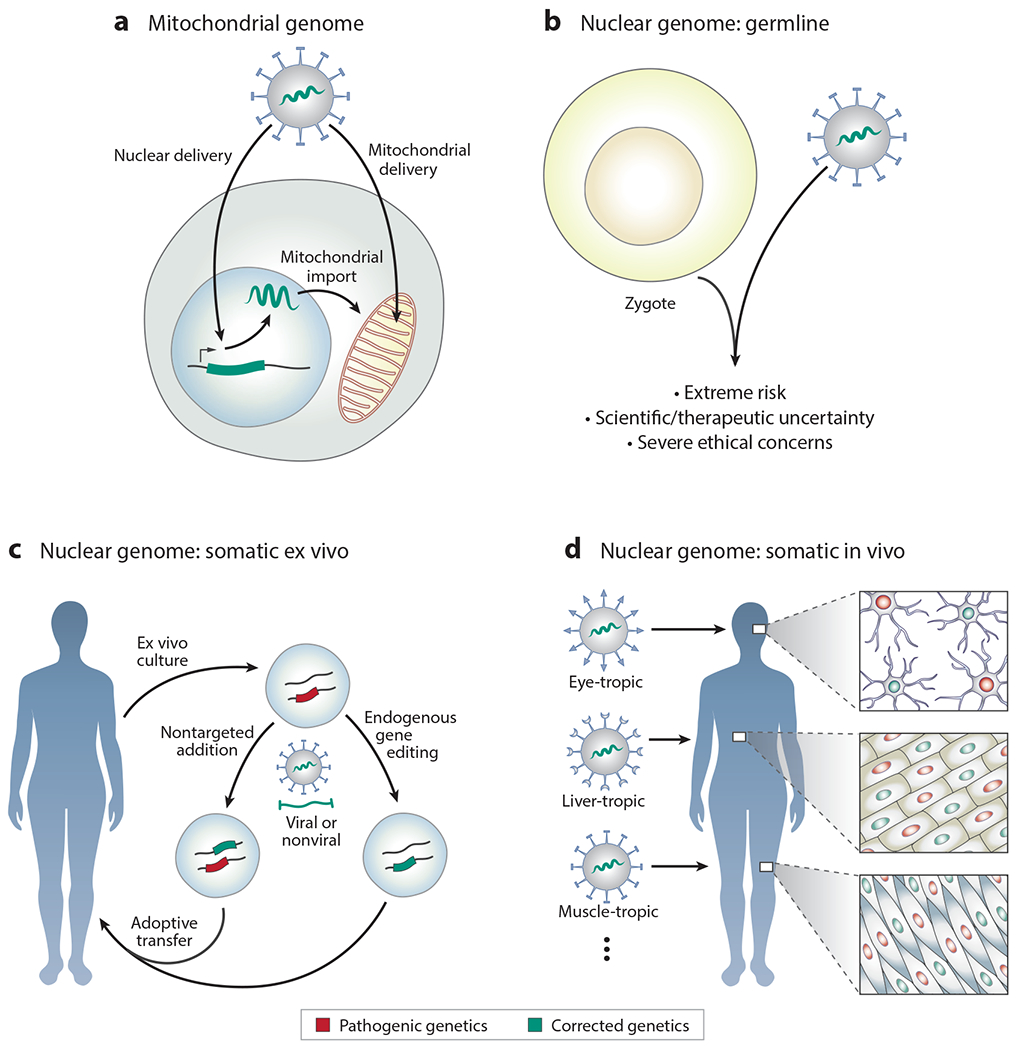
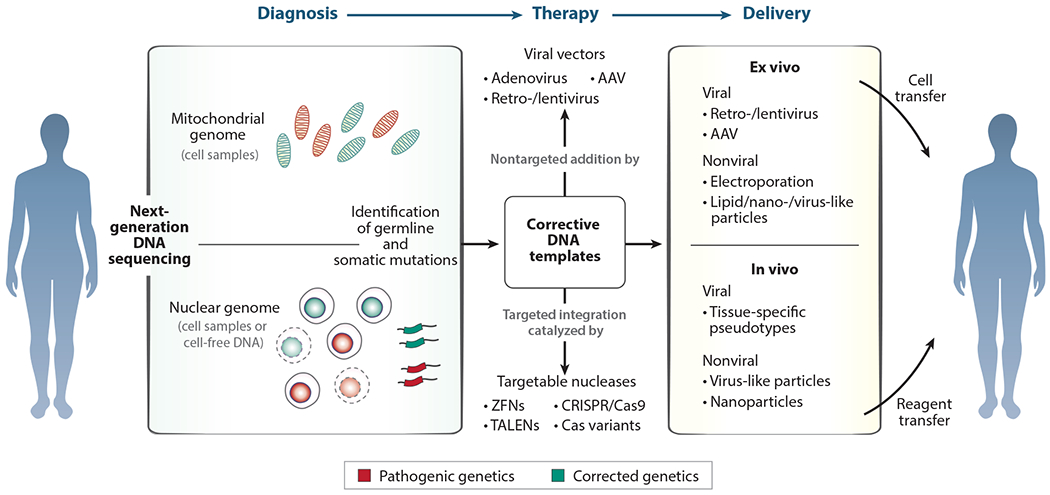
References
-
- Motulsky AG. 2010. History of human genetics. In Vogel and Motulsky’s Human Genetics: Problems and Approaches, ed. Speicher MR, Motulsky AG, Antonarakis SE, pp. 13–29. Heidelberg, Ger.: Springer. 4th ed.
-
- Soler A, Morales C, Mademont-Soler I, Margarit E, Borrell A, et al. 2017. Overview of chromosome abnormalities in first trimester miscarriages: a series of 1,011 consecutive chorionic villi sample karyotypes. Cytogenet. Genome Res 152:81–89 - PubMed
-
- Ingram VM. 1956. A specific chemical difference between the globins of normal human and sickle-cell anaemia haemoglobin. Nature 178:792–94 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
