

17β-Estradiol and estrogen receptor α protect right ventricular function in pulmonary hypertension via BMPR2 and apelin
- PMID: 33497359
- PMCID: PMC7968046
- DOI: 10.1172/JCI129433
17β-Estradiol and estrogen receptor α protect right ventricular function in pulmonary hypertension via BMPR2 and apelin
Abstract
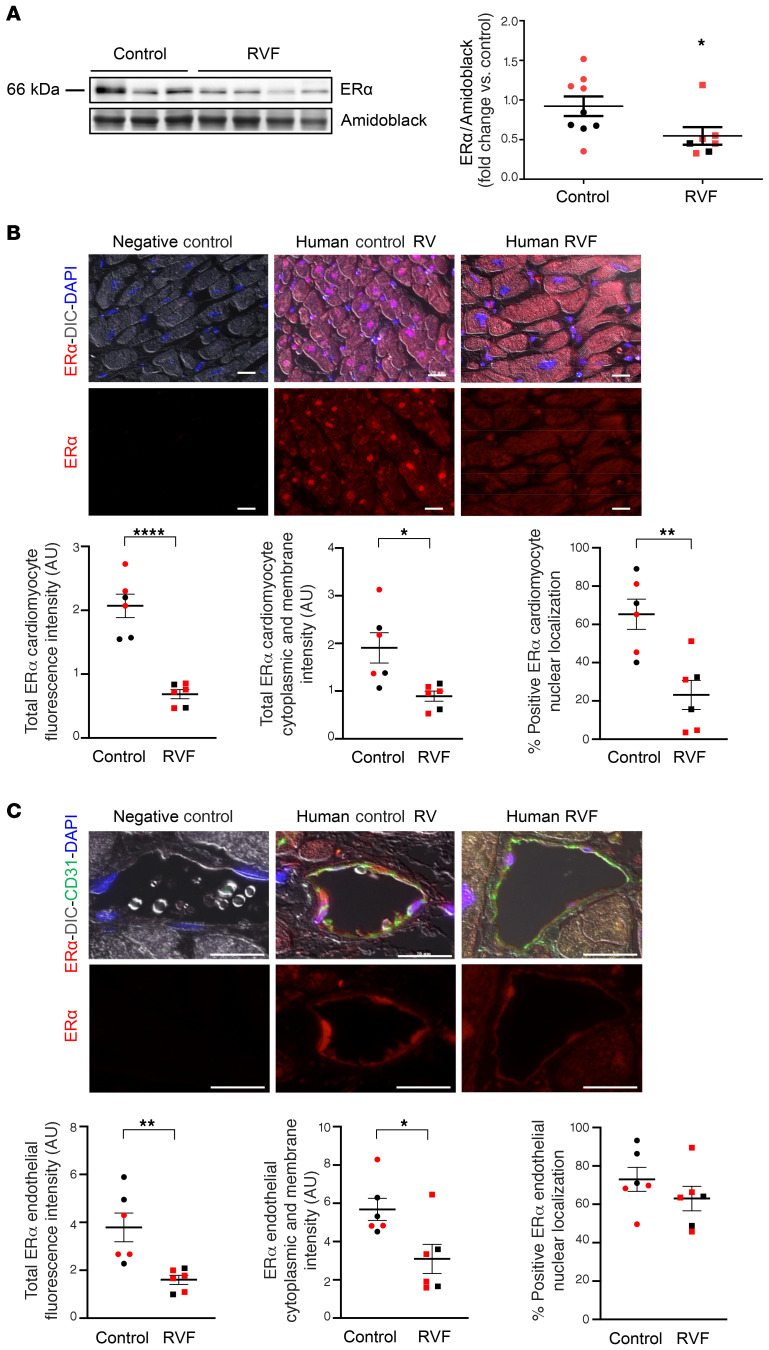
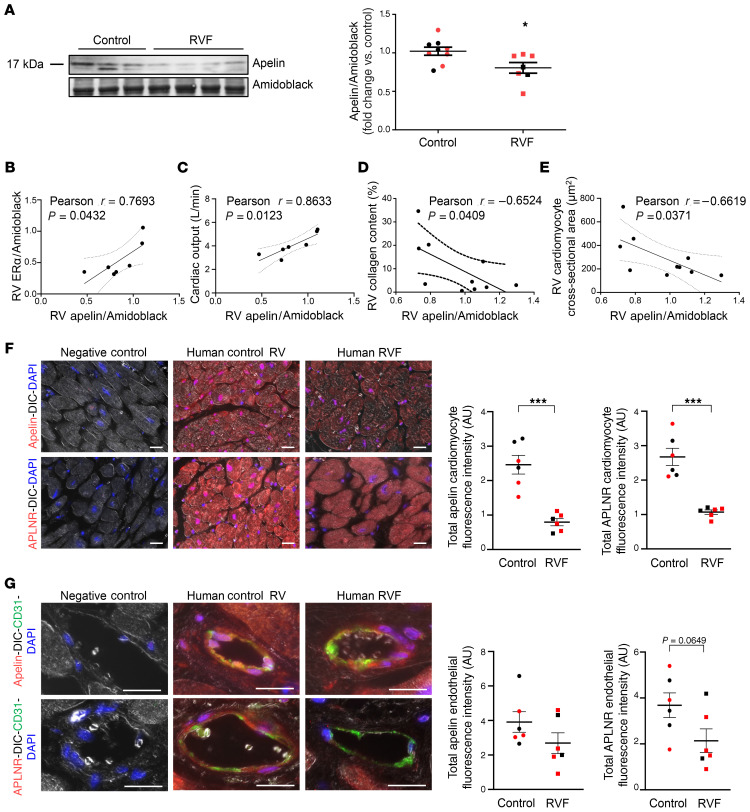
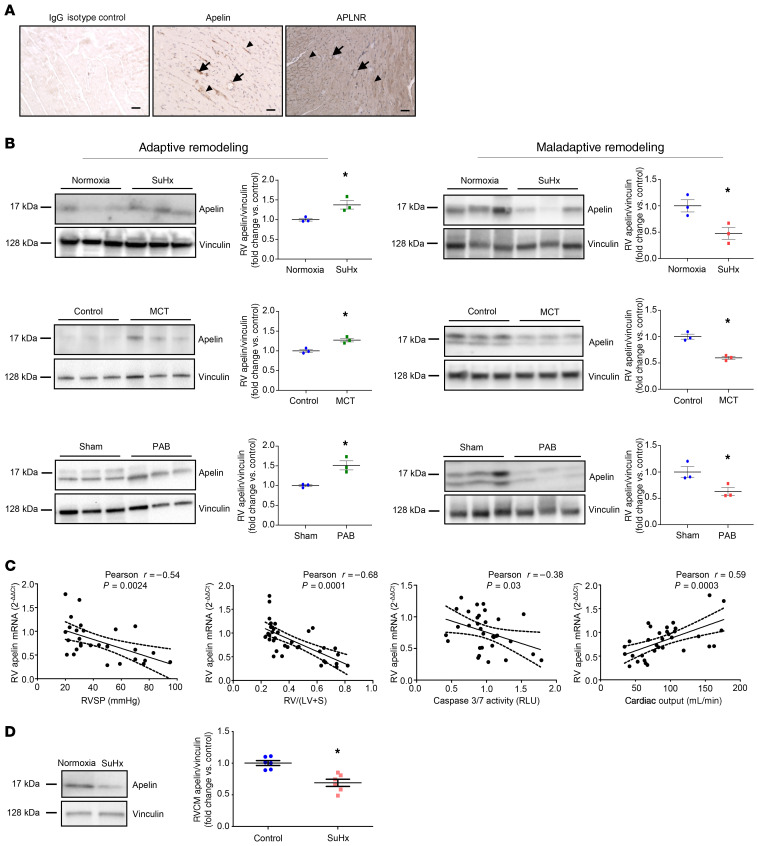
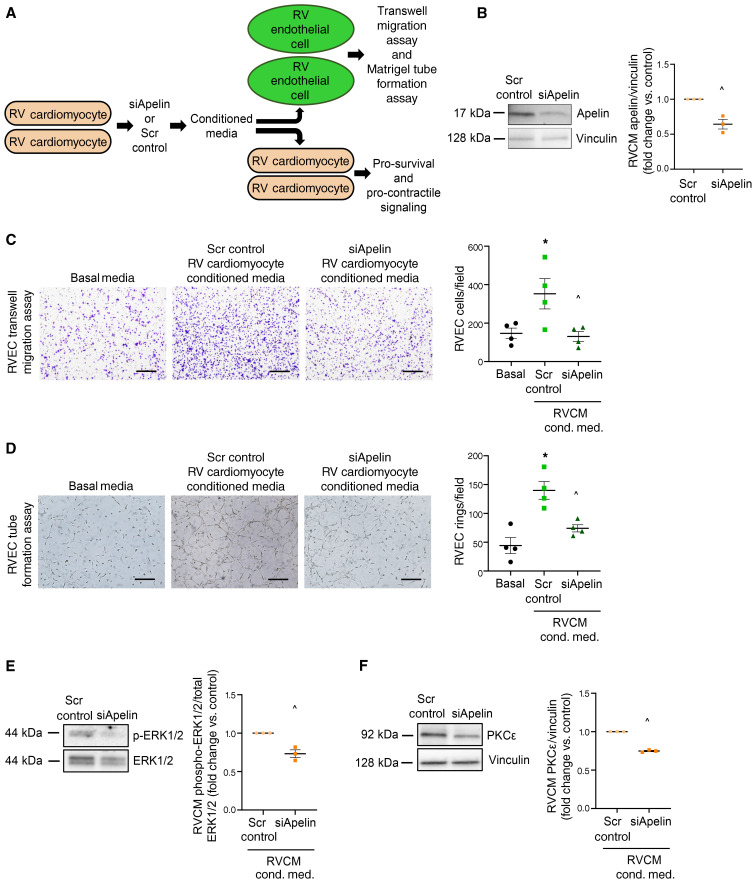
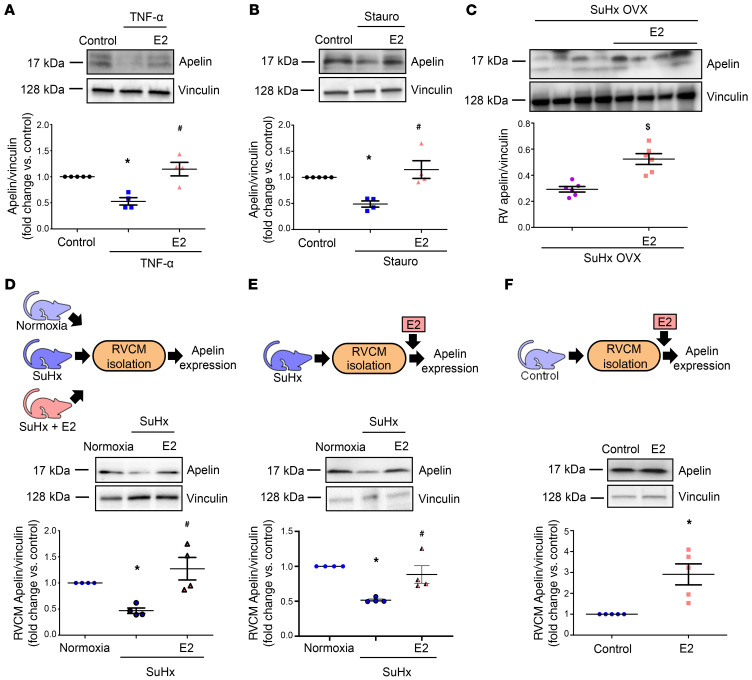
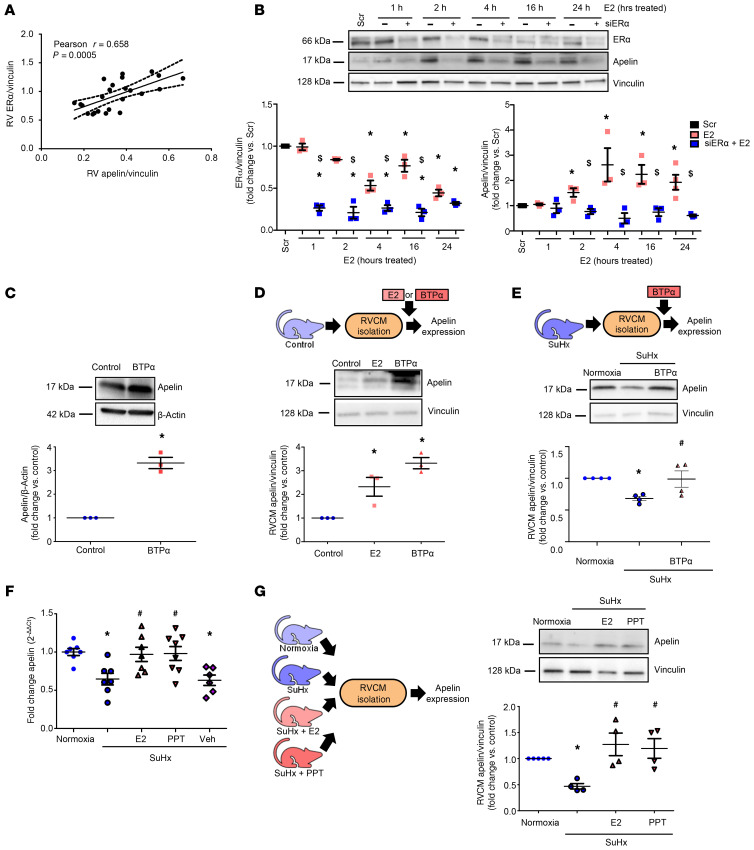
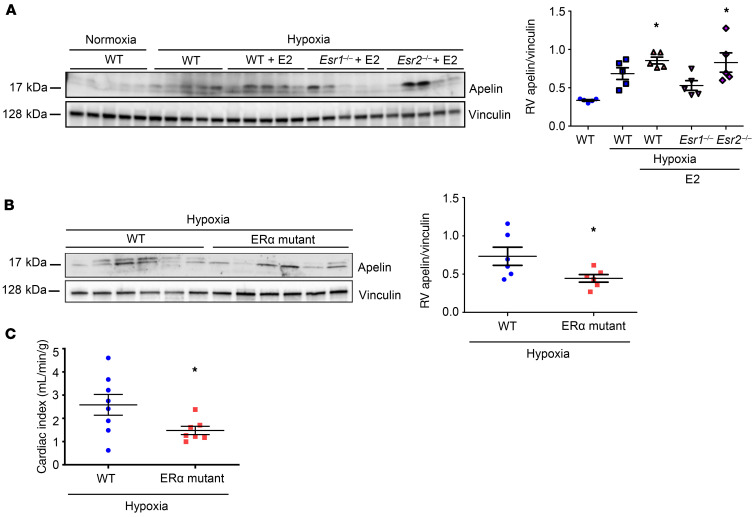
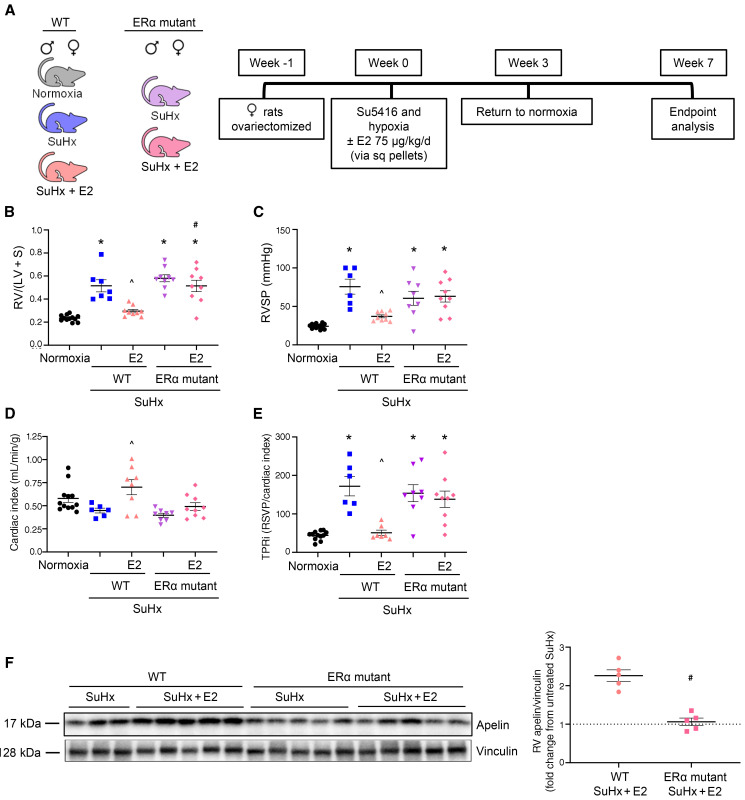
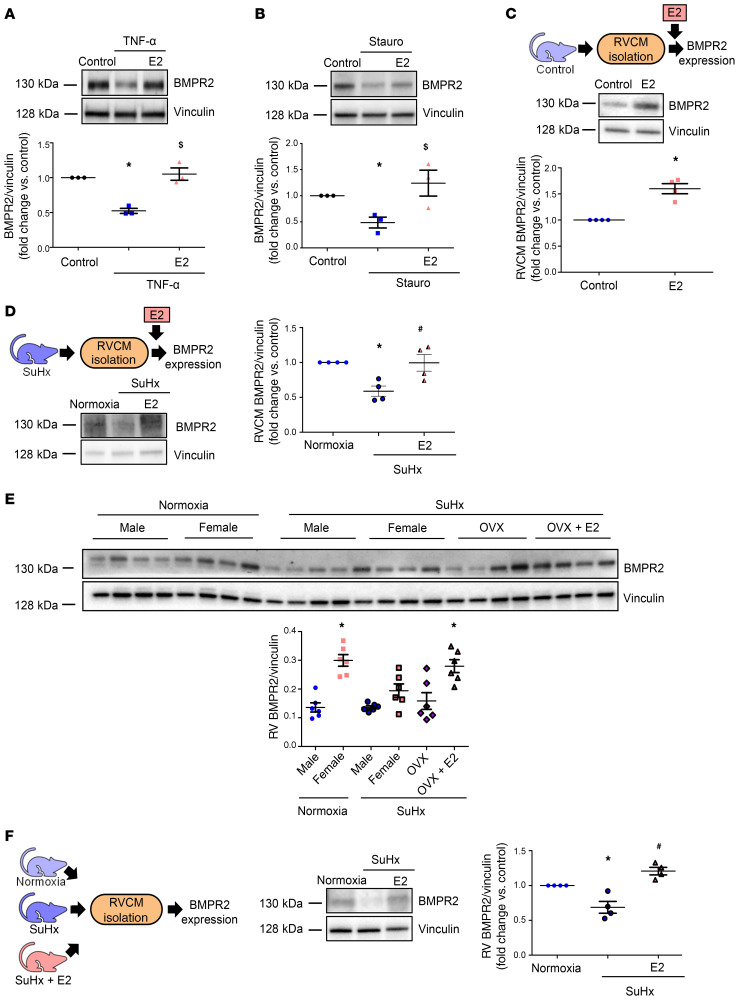
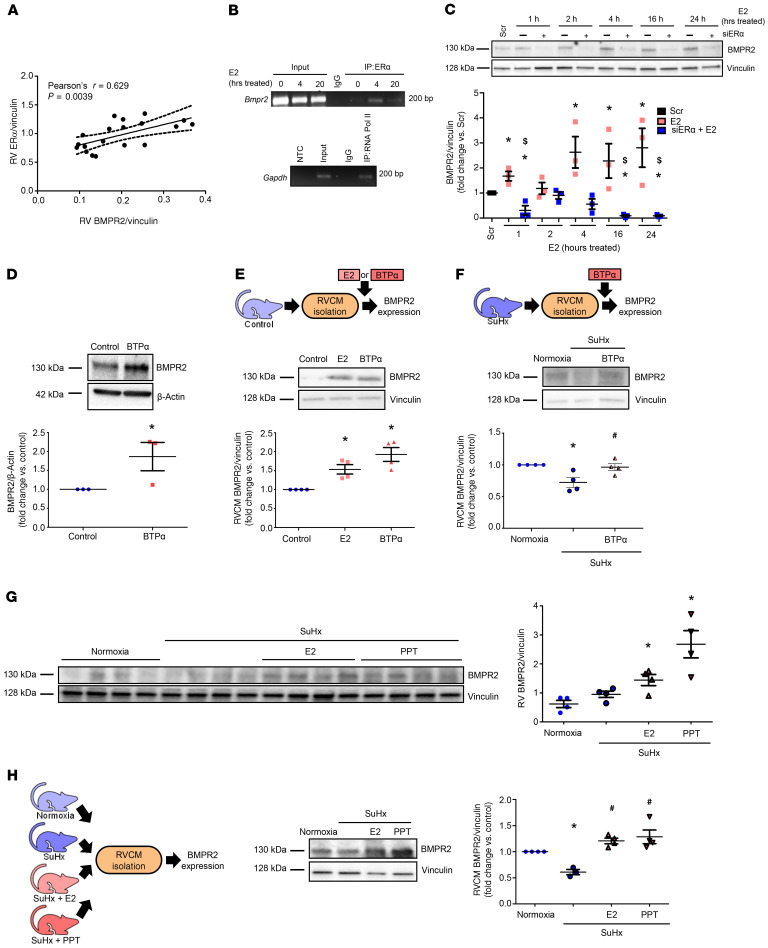
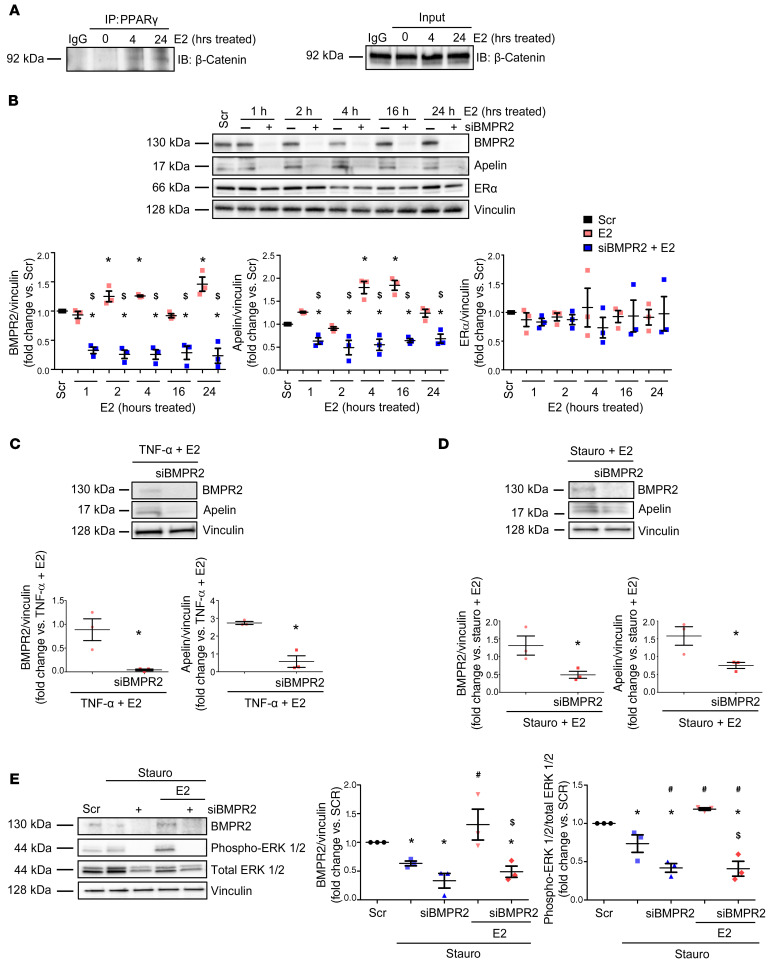
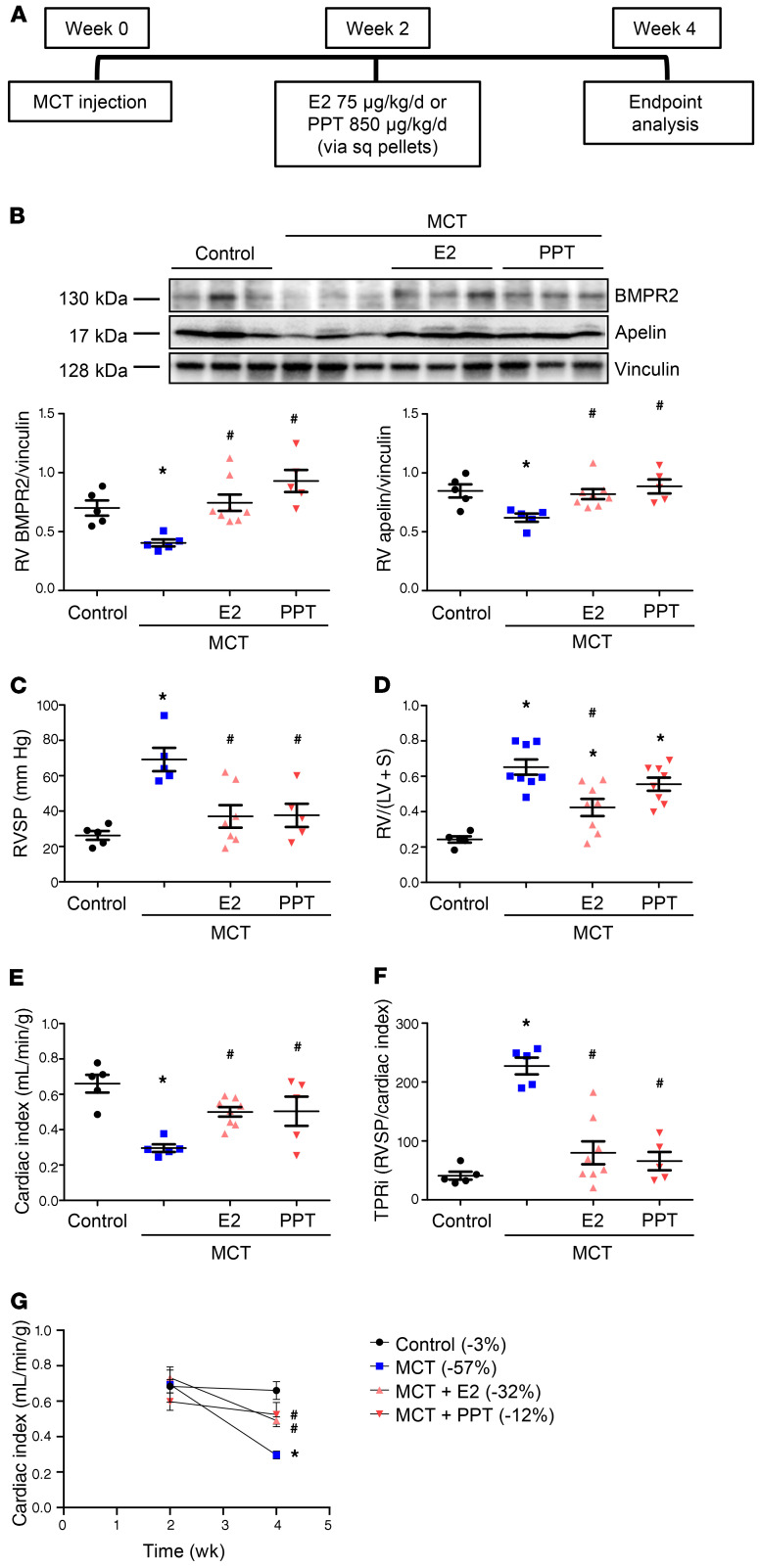
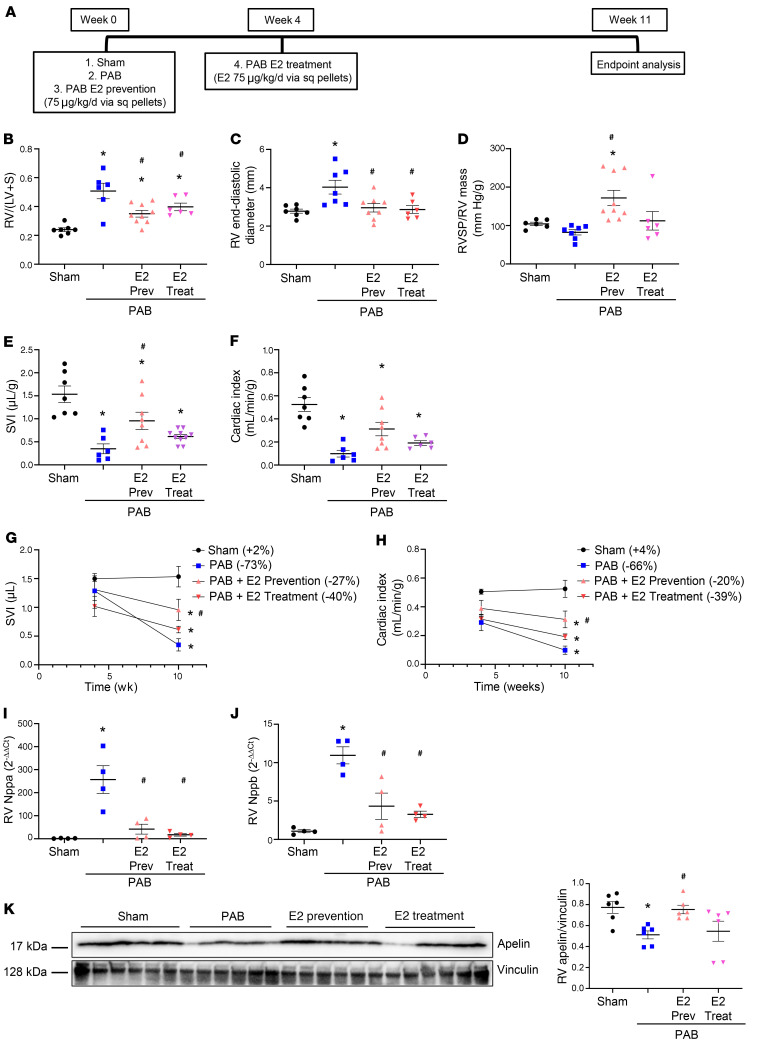
Women with pulmonary arterial hypertension (PAH) exhibit better right ventricular (RV) function and survival than men; however, the underlying mechanisms are unknown. We hypothesized that 17β-estradiol (E2), through estrogen receptor α (ER-α), attenuates PAH-induced RV failure (RVF) by upregulating the procontractile and prosurvival peptide apelin via a BMPR2-dependent mechanism. We found that ER-α and apelin expression were decreased in RV homogenates from patients with RVF and from rats with maladaptive (but not adaptive) RV remodeling. RV cardiomyocyte apelin abundance increased in vivo or in vitro after treatment with E2 or ER-α agonist. Studies employing ER-α-null or ER-β-null mice, ER-α loss-of-function mutant rats, or siRNA demonstrated that ER-α is necessary for E2 to upregulate RV apelin. E2 and ER-α increased BMPR2 in pulmonary hypertension RVs and in isolated RV cardiomyocytes, associated with ER-α binding to the Bmpr2 promoter. BMPR2 is required for E2-mediated increases in apelin abundance, and both BMPR2 and apelin are necessary for E2 to exert RV-protective effects. E2 or ER-α agonist rescued monocrotaline pulmonary hypertension and restored RV apelin and BMPR2. We identified what we believe to be a novel cardioprotective E2/ER-α/BMPR2/apelin axis in the RV. Harnessing this axis may lead to novel RV-targeted therapies for PAH patients of either sex.
Keywords: Heart failure; Molecular biology; Pulmonology; Sex hormones; Vascular Biology.
Conflict of interest statement
Figures
Comment in
-
Role of sex hormones in cardiovascular diseases.Nat Rev Cardiol. 2021 Jun;18(6):385. doi: 10.1038/s41569-021-00551-2. Nat Rev Cardiol. 2021. PMID: 33828253 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
