

Chemotherapy Induces Senescence-Like Resilient Cells Capable of Initiating AML Recurrence
- PMID: 33500244
- PMCID: PMC8178167
- DOI: 10.1158/2159-8290.CD-20-1375
Chemotherapy Induces Senescence-Like Resilient Cells Capable of Initiating AML Recurrence
Abstract
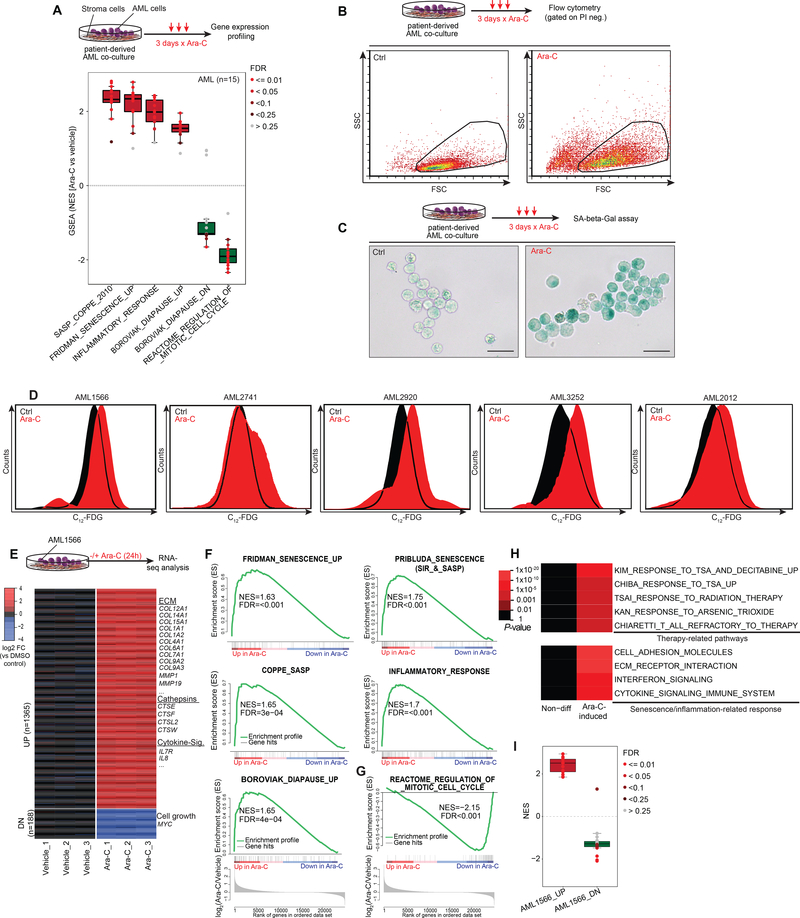
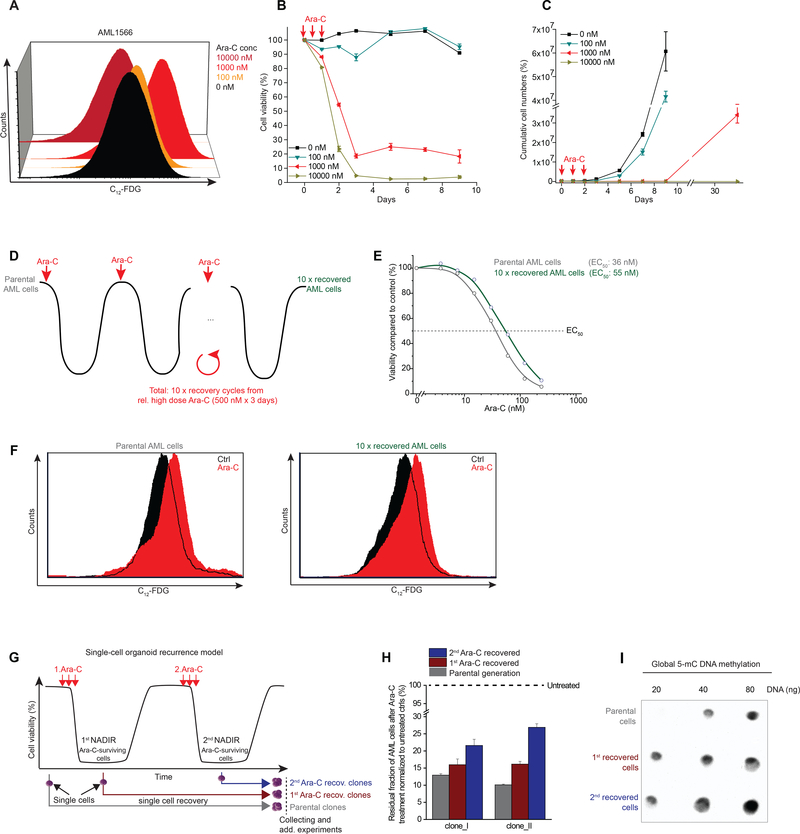
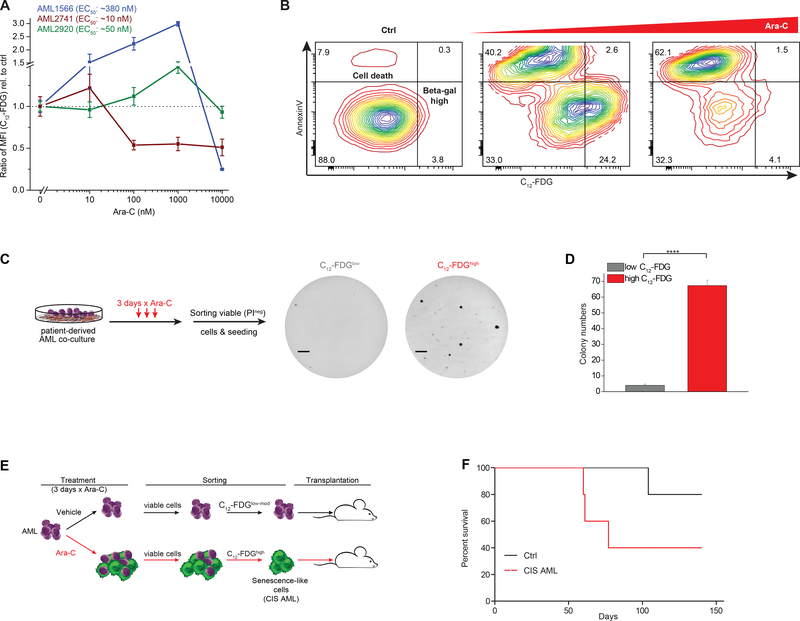
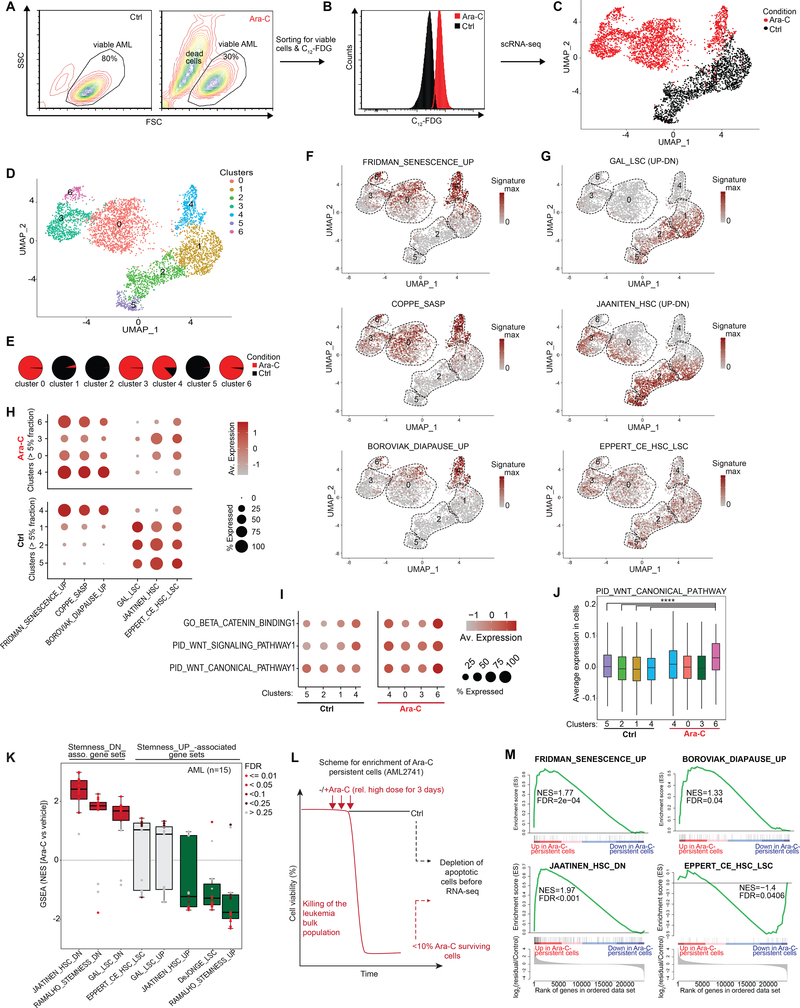
Patients with acute myeloid leukemia (AML) frequently relapse after chemotherapy, yet the mechanism by which AML reemerges is not fully understood. Herein, we show that primary AML cells enter a senescence-like phenotype following chemotherapy in vitro and in vivo. This is accompanied by induction of senescence/inflammatory and embryonic diapause transcriptional programs, with downregulation of MYC and leukemia stem cell genes. Single-cell RNA sequencing suggested depletion of leukemia stem cells in vitro and in vivo, and enrichment for subpopulations with distinct senescence-like cells. This senescence effect was transient and conferred superior colony-forming and engraftment potential. Entry into this senescence-like phenotype was dependent on ATR, and persistence of AML cells was severely impaired by ATR inhibitors. Altogether, we propose that AML relapse is facilitated by a senescence-like resilience phenotype that occurs regardless of their stem cell status. Upon recovery, these post-senescence AML cells give rise to relapsed AMLs with increased stem cell potential. SIGNIFICANCE: Despite entering complete remission after chemotherapy, relapse occurs in many patients with AML. Thus, there is an urgent need to understand the relapse mechanism in AML and the development of targeted treatments to improve outcome. Here, we identified a senescence-like resilience phenotype through which AML cells can survive and repopulate leukemia.This article is highlighted in the In This Issue feature, p. 1307.
©2021 American Association for Cancer Research.
Figures
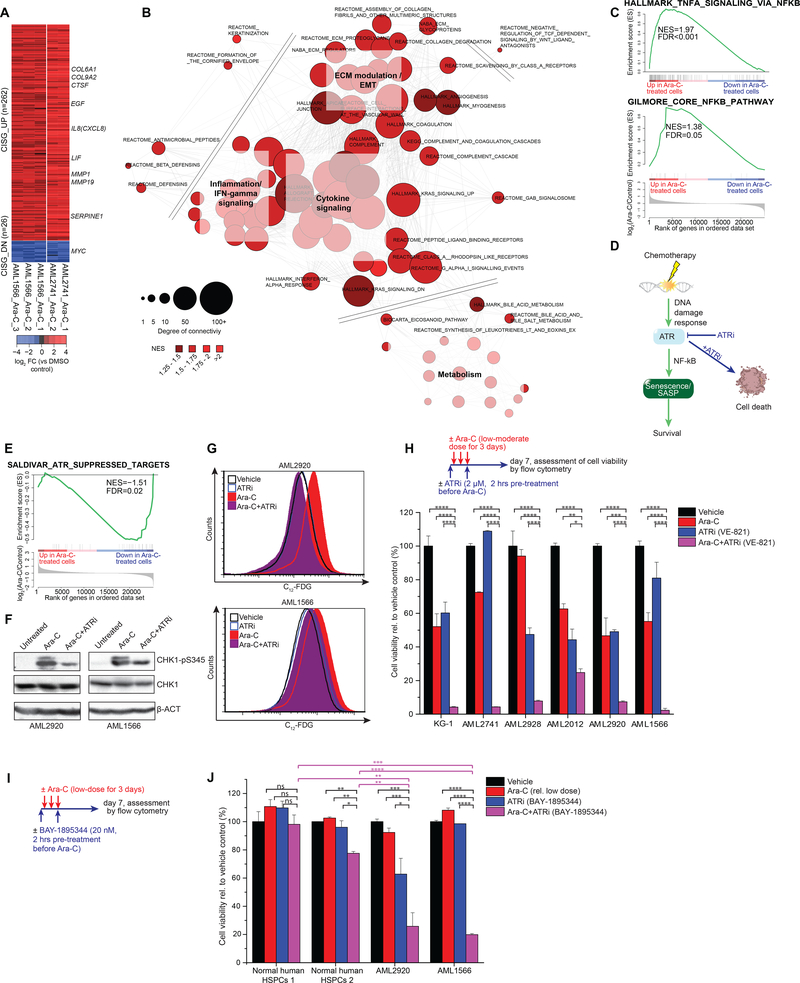
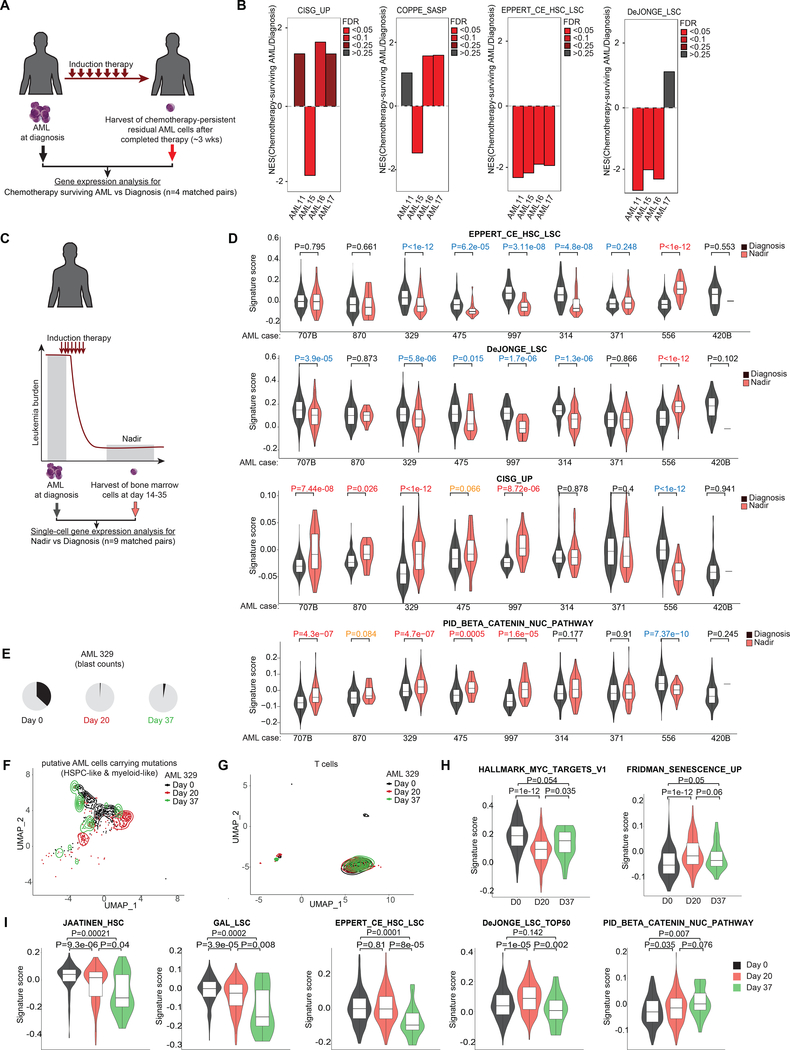
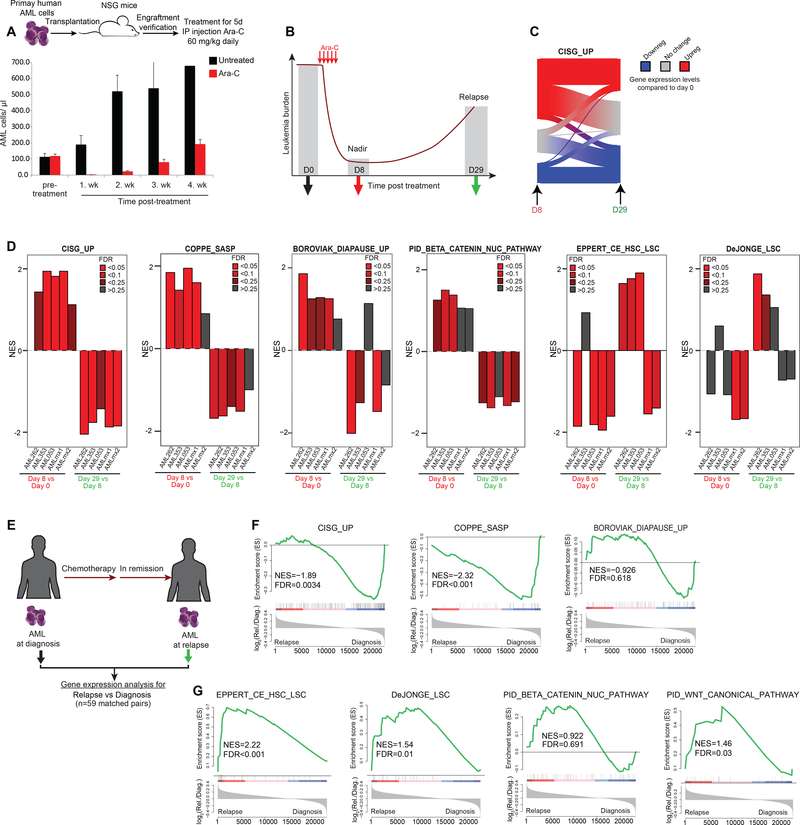
References
-
- Burnett A, Wetzler M, Lowenberg B. Therapeutic advances in acute myeloid leukemia. JClinOncol 2011;29(5):487–94. - PubMed
-
- Farge T, Saland E, de Toni F, Aroua N, Hosseini M, Perry R, et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer discovery 2017;7(7):716–35 doi 10.1158/2159-8290.CD-16-0441. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
