

Single-cell proteomic and transcriptomic analysis of macrophage heterogeneity using SCoPE2
- PMID: 33504367
- PMCID: PMC7839219
- DOI: 10.1186/s13059-021-02267-5
Single-cell proteomic and transcriptomic analysis of macrophage heterogeneity using SCoPE2
Abstract
Background: Macrophages are innate immune cells with diverse functional and molecular phenotypes. This diversity is largely unexplored at the level of single-cell proteomes because of the limitations of quantitative single-cell protein analysis.
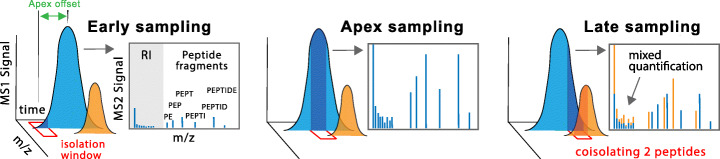
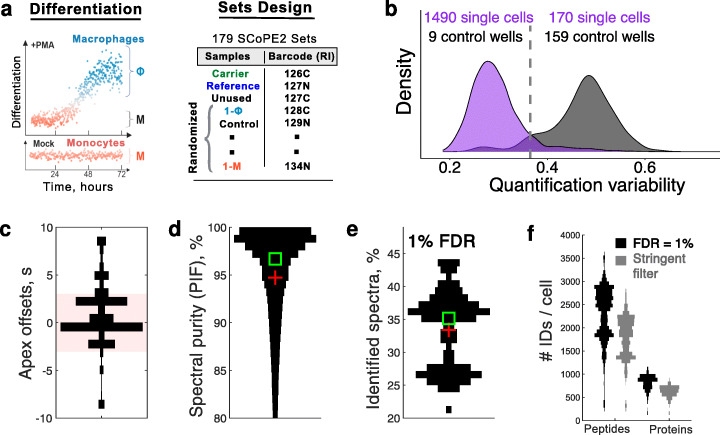
Results: To overcome this limitation, we develop SCoPE2, which substantially increases quantitative accuracy and throughput while lowering cost and hands-on time by introducing automated and miniaturized sample preparation. These advances enable us to analyze the emergence of cellular heterogeneity as homogeneous monocytes differentiate into macrophage-like cells in the absence of polarizing cytokines. SCoPE2 quantifies over 3042 proteins in 1490 single monocytes and macrophages in 10 days of instrument time, and the quantified proteins allow us to discern single cells by cell type. Furthermore, the data uncover a continuous gradient of proteome states for the macrophages, suggesting that macrophage heterogeneity may emerge in the absence of polarizing cytokines. Parallel measurements of transcripts by 10× Genomics suggest that our measurements sample 20-fold more protein copies than RNA copies per gene, and thus, SCoPE2 supports quantification with improved count statistics. This allowed exploring regulatory interactions, such as interactions between the tumor suppressor p53, its transcript, and the transcripts of genes regulated by p53.
Conclusions: Even in a homogeneous environment, macrophage proteomes are heterogeneous. This heterogeneity correlates to the inflammatory axis of classically and alternatively activated macrophages. Our methodology lays the foundation for automated and quantitative single-cell analysis of proteins by mass spectrometry and demonstrates the potential for inferring transcriptional and post-transcriptional regulation from variability across single cells.
Conflict of interest statement
The authors declare that they have no competing financial interests.
Correspondence: Correspondence and materials requests should be addressed to nslavov@alum.mit.edu
Figures





Comment in
-
Towards resolving proteomes in single cells.Nat Methods. 2021 Aug;18(8):856. doi: 10.1038/s41592-021-01243-y. Nat Methods. 2021. PMID: 34354289 No abstract available.
References
-
- Ben-Moshe S, Itzkovitz S. Spatial heterogeneity in the mammalian liver. Nature Reviews Gastroenterol Hepatol. 2019;16(7):395–410. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
