

RHOBTB2 Mutations Expand the Phenotypic Spectrum of Alternating Hemiplegia of Childhood
- PMID: 33504645
- PMCID: PMC8032376
- DOI: 10.1212/WNL.0000000000011543
RHOBTB2 Mutations Expand the Phenotypic Spectrum of Alternating Hemiplegia of Childhood
Abstract
Objective: To explore the phenotypic spectrum of RHOBTB2-related disorders and specifically to determine whether patients fulfill criteria for alternating hemiplegia of childhood (AHC), we report the clinical features of 11 affected individuals.
Methods: Individuals with RHOBTB2-related disorders were identified through a movement disorder clinic at a specialist pediatric center, with additional cases identified through collaboration with other centers internationally. Clinical data were acquired through retrospective case-note review.
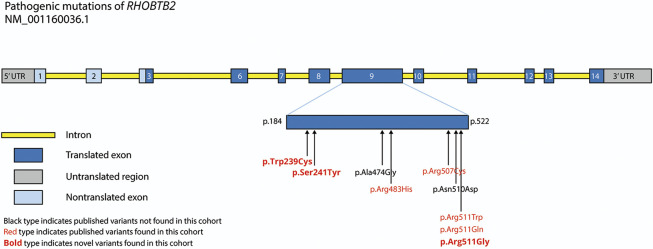
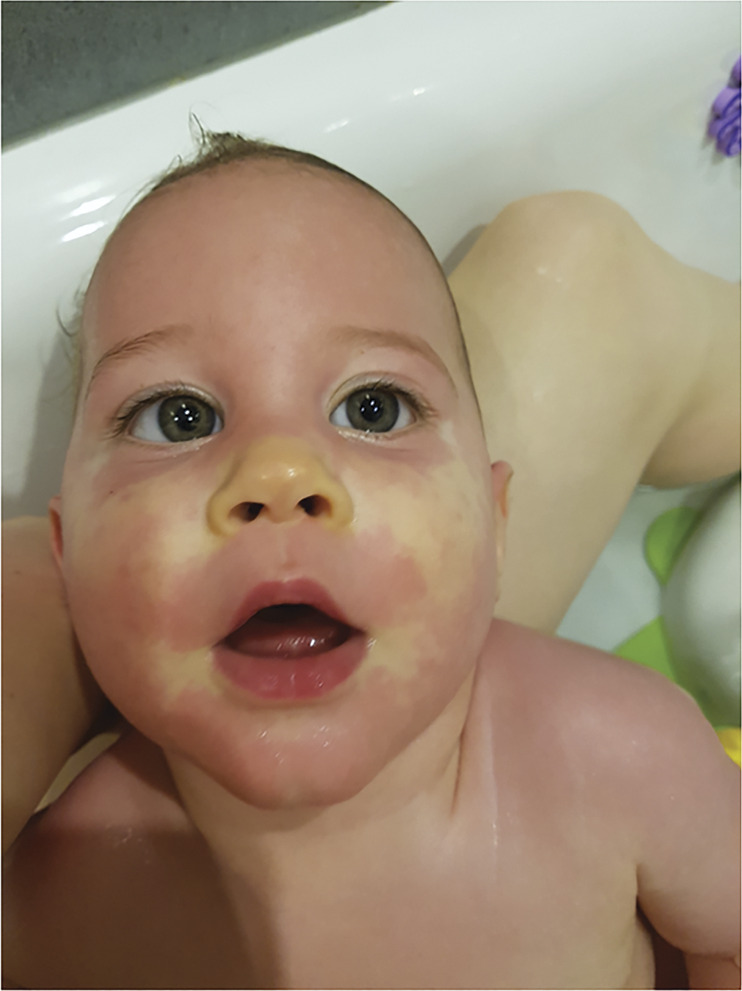
Results: Eleven affected patients were identified. All had heterozygous missense variants involving exon 9 of RHOBTB2, confirmed as de novo in 9 cases. All had a complex motor phenotype, including at least 2 different kinds of movement disorder, e.g., ataxia and dystonia. Many patients demonstrated several features fulfilling the criteria for AHC: 10 patients had a movement disorder including paroxysmal elements, and 8 experienced hemiplegic episodes. In contrast to classic AHC, commonly caused by mutations in ATP1A3, these events were reported later only in RHOBTB2 mutation-positive patients from 20 months of age. Seven patients had epilepsy, but of these, 4 patients achieved seizure freedom. All patients had intellectual disability, usually moderate to severe. Other features include episodes of marked skin color change and gastrointestinal symptoms, each in 4 patients.
Conclusion: Although heterozygous RHOBTB2 mutations were originally described in early infantile epileptic encephalopathy type 64, our study confirms that they account for a more expansive clinical phenotype, including a complex polymorphic movement disorder with paroxysmal elements resembling AHC. RHOBTB2 testing should therefore be considered in patients with an AHC-like phenotype, particularly those negative for ATPA1A3 mutations.
© 2021 American Academy of Neurology.
Figures
References
-
- Lopes F, Barbosa M, Ameur A, et al. Identification of novel genetic causes of Rett syndrome-like phenotypes. J Med Genet 2016;53:190–199. - PubMed
-
- Belal H, Nakashima M, Matsumoto H. De novo variants in RHOBTB2, an atypical Rho GTPase gene, cause epileptic encephalopathy. Hum Mutat 2018;39:1070–1075. - PubMed
-
- IHS Classification ICHD-3. London: International Headache Society; 2019. Available at: ichd-3.org/appendix/a1-migraine/a1-6-episodic-syndromes-that-may-be-asso.... Accessed September 2019.
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases