

Impaired prenatal motor axon development necessitates early therapeutic intervention in severe SMA
- PMID: 33504650
- PMCID: PMC8208236
- DOI: 10.1126/scitranslmed.abb6871
Impaired prenatal motor axon development necessitates early therapeutic intervention in severe SMA
Abstract
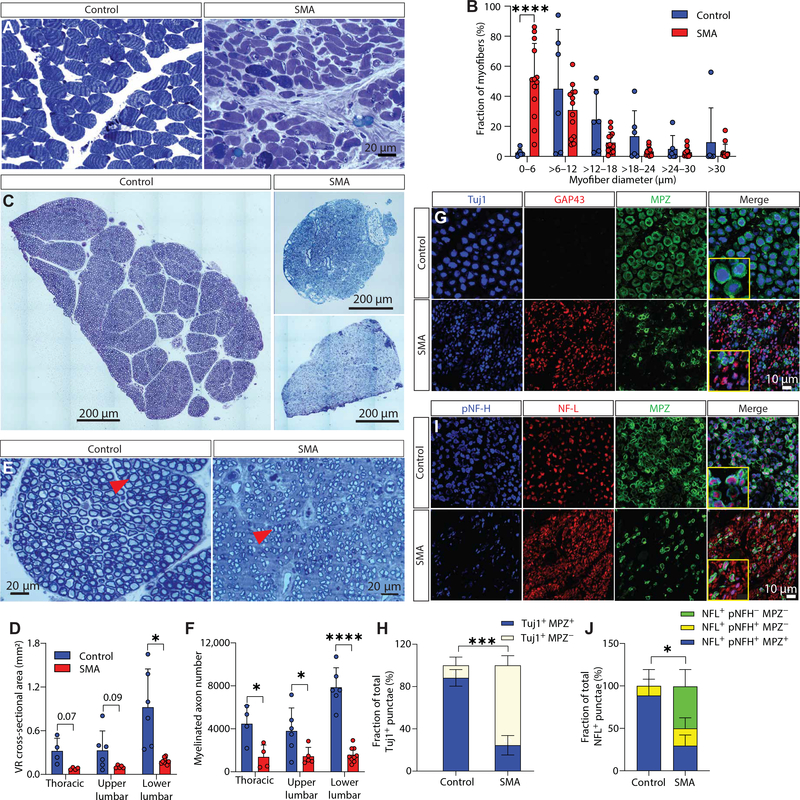
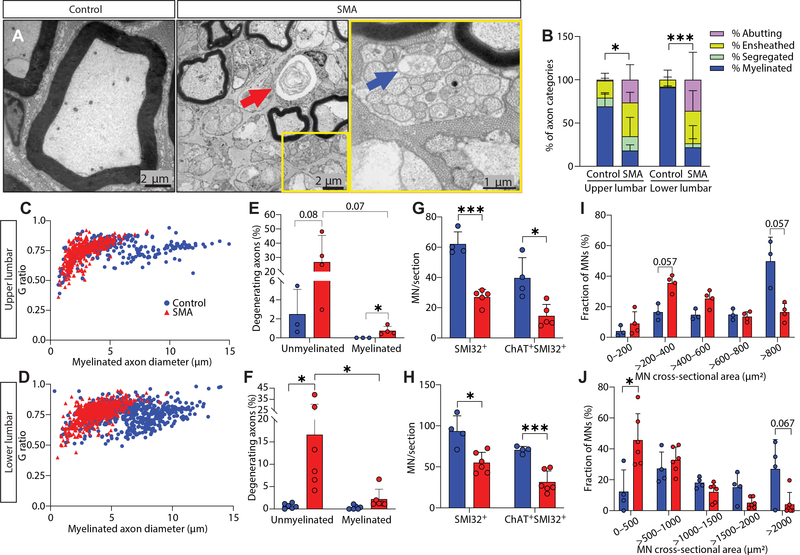
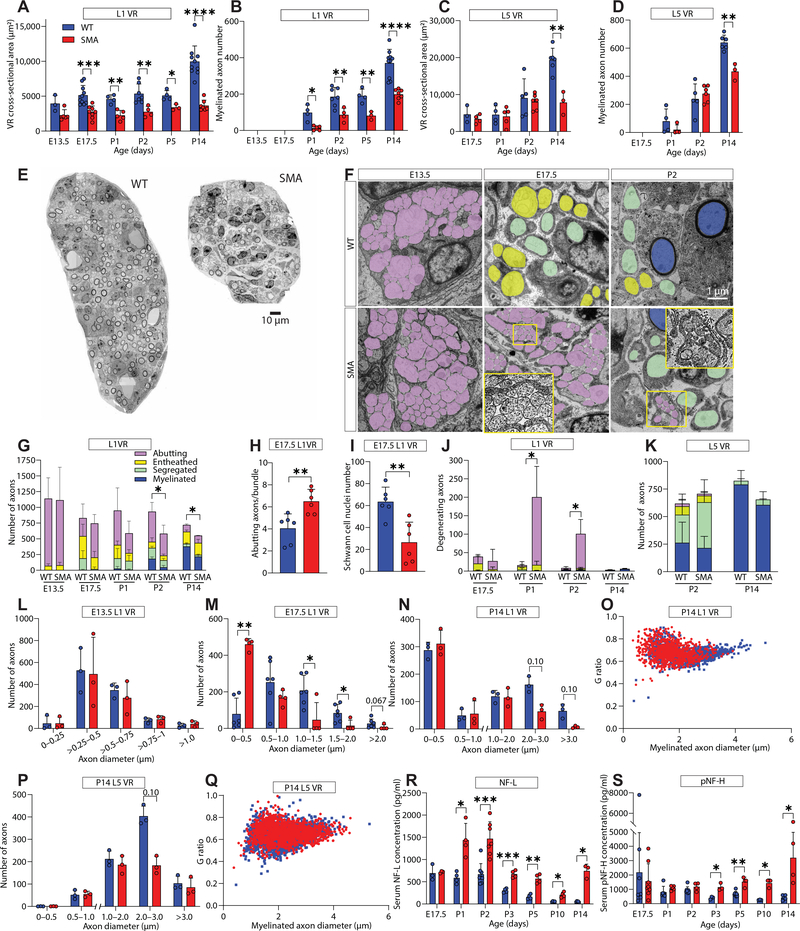
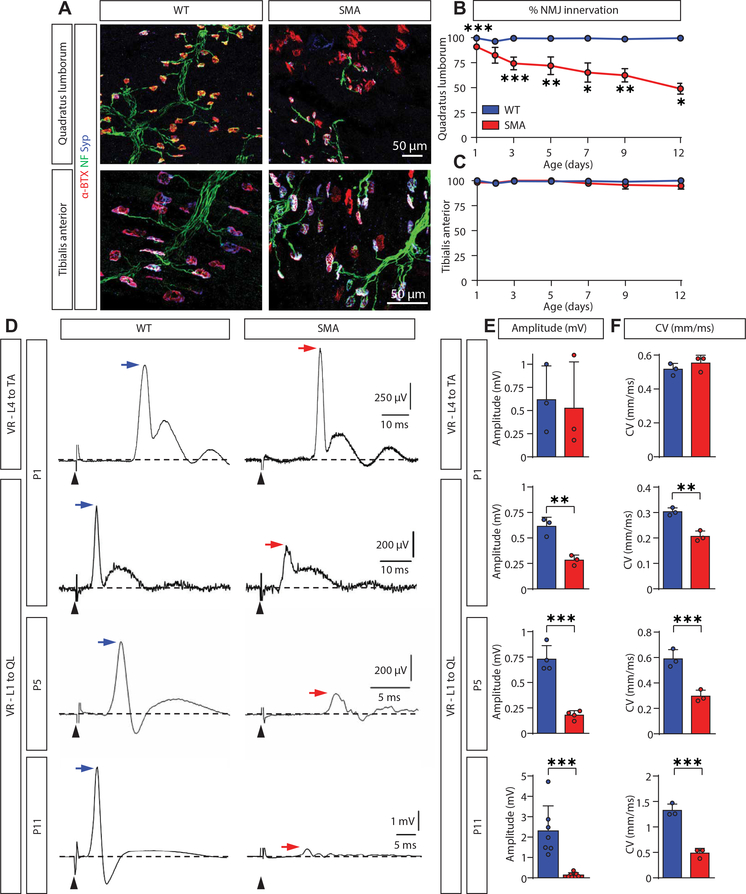
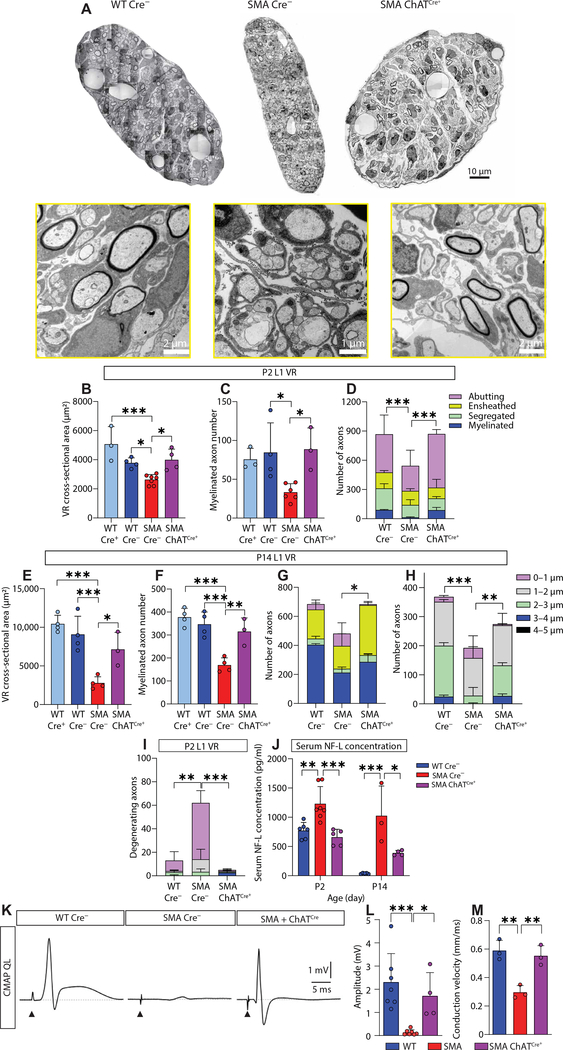
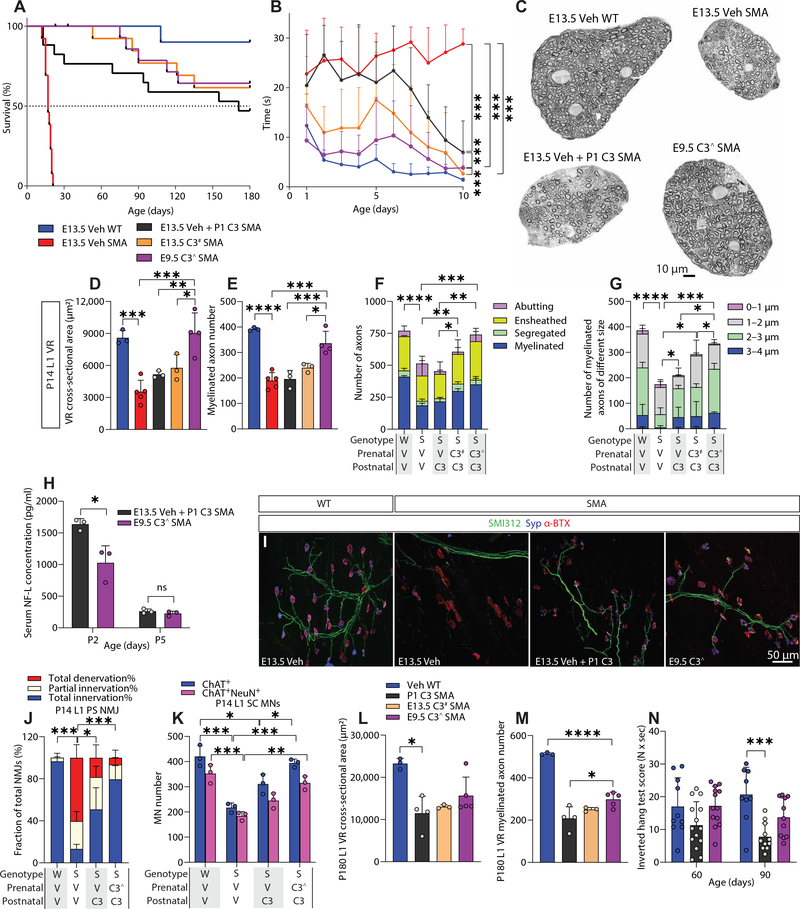
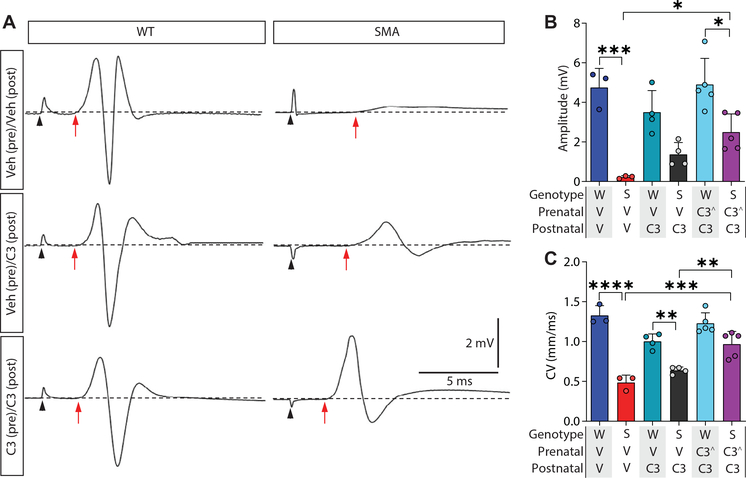
Gene replacement and pre-mRNA splicing modifier therapies represent breakthrough gene targeting treatments for the neuromuscular disease spinal muscular atrophy (SMA), but mechanisms underlying variable efficacy of treatment are incompletely understood. Our examination of severe infantile onset human SMA tissues obtained at expedited autopsy revealed persistence of developmentally immature motor neuron axons, many of which are actively degenerating. We identified similar features in a mouse model of severe SMA, in which impaired radial growth and Schwann cell ensheathment of motor axons began during embryogenesis and resulted in reduced acquisition of myelinated axons that impeded motor axon function neonatally. Axons that failed to ensheath degenerated rapidly postnatally, specifically releasing neurofilament light chain protein into the blood. Genetic restoration of survival motor neuron protein (SMN) expression in mouse motor neurons, but not in Schwann cells or muscle, improved SMA motor axon development and maintenance. Treatment with small-molecule SMN2 splice modifiers beginning immediately after birth in mice increased radial growth of the already myelinated axons, but in utero treatment was required to restore axonal growth and associated maturation, prevent subsequent neonatal axon degeneration, and enhance motor axon function. Together, these data reveal a cellular basis for the fulminant neonatal worsening of patients with infantile onset SMA and identify a temporal window for more effective treatment. These findings suggest that minimizing treatment delay is critical to achieve optimal therapeutic efficacy.
Copyright © 2021 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Figures
References
-
- Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, Le Paslier D, Frézal J, Cohen D, Weissenbach J, Munnich A, Melki J, Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80, 155–165 (1995). - PubMed
-
- Monani UR, Lorson CL, Parsons DW, Prior TW, Androphy EJ, Burghes AH, McPherson JD, A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 8, 1177–1183 (1999). - PubMed
-
- Finkel RS, McDermott MP, Kaufmann P, Darras BT, Chung WK, Sproule DM, Kang PB, Foley AR, Yang ML, Martens WB, Oskoui M, Glanzman AM, Flickinger J, Montes J, Dunaway S, O’Hagen J, Quigley J, Riley S, Benton M, Ryan PA, Montgomery M, Marra J, Gooch C, De Vivo DC, Observational study of spinal muscular atrophy type I and implications for clinical trials. Neurology 83, 810–817 (2014). - PMC - PubMed
-
- Kolb SJ, Coffey CS, Yankey JW, Krosschell K, Arnold WD, Rutkove SB, Swoboda KJ, Reyna SP, Sakonju A, Darras BT, Shell R, Kuntz N, Castro D, Parsons J, Connolly AM, Chiriboga CA, McDonald C, Burnette WB, Werner K, Thangarajh M, Shieh PB, Finanger E, Cudkowicz ME, McGovern MM, McNeil DE, Finkel R, Iannaccone ST, Kaye E, Kingsley A, Renusch SR, McGovern VL, Wang X, Zaworski PG, Prior TW, Burghes AHM, Bartlett A, Kissel JT; NeuroNEXT Clinical Trial Network on behalf of the NN101 SMA Biomarker Investigators, Natural history of infantile-onset spinal muscular atrophy. Ann. Neurol. 82, 883–891 (2017). - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
