

Mapping the immune environment in clear cell renal carcinoma by single-cell genomics
- PMID: 33504936
- PMCID: PMC7840906
- DOI: 10.1038/s42003-020-01625-6
Mapping the immune environment in clear cell renal carcinoma by single-cell genomics
Abstract
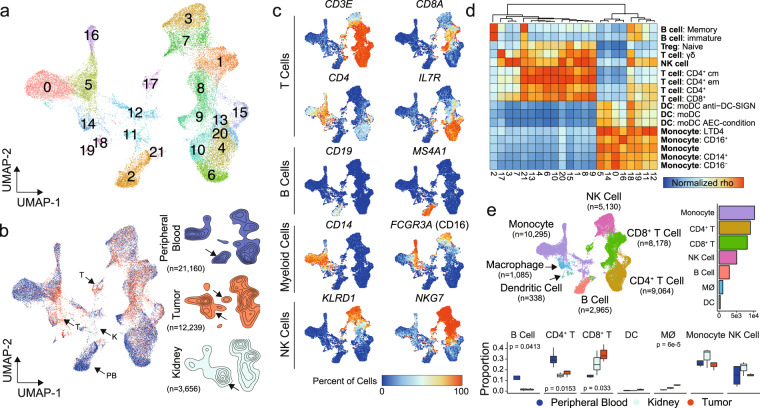
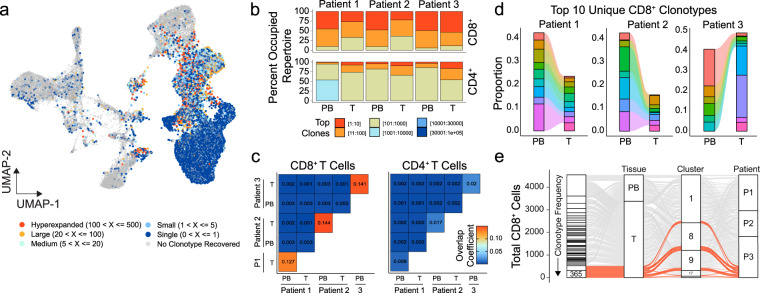
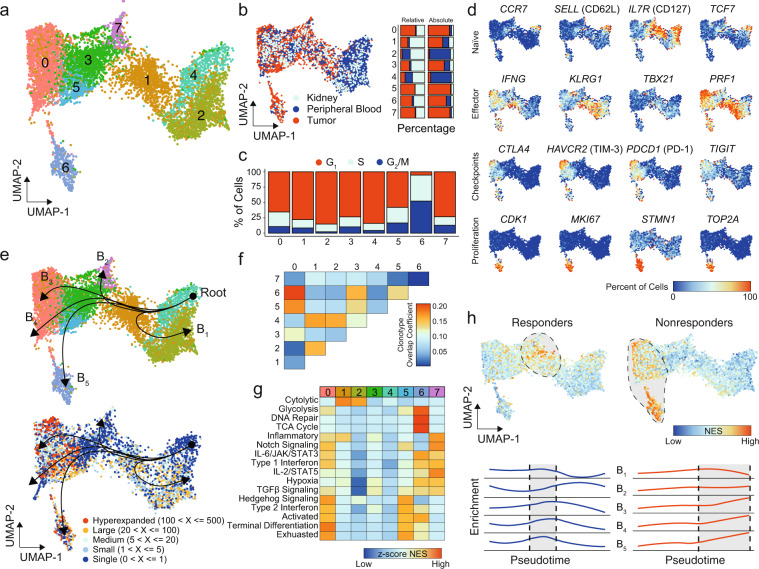
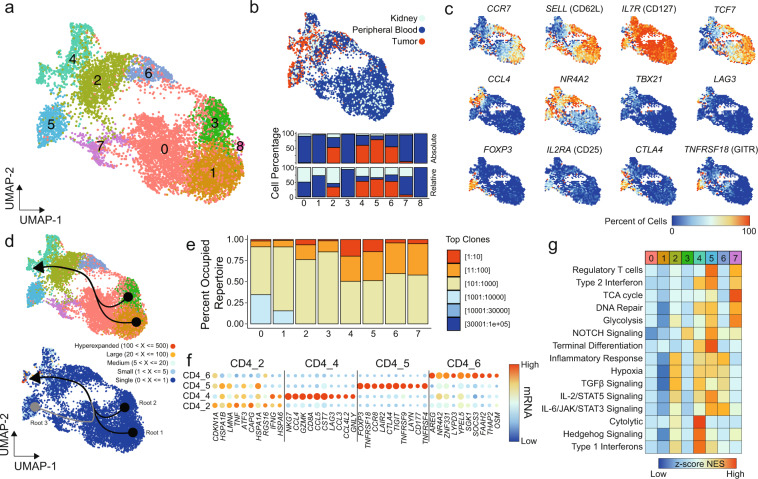
Clear cell renal cell carcinoma (ccRCC) is one of the most immunologically distinct tumor types due to high response rate to immunotherapies, despite low tumor mutational burden. To characterize the tumor immune microenvironment of ccRCC, we applied single-cell-RNA sequencing (SCRS) along with T-cell-receptor (TCR) sequencing to map the transcriptomic heterogeneity of 25,688 individual CD45+ lymphoid and myeloid cells in matched tumor and blood from three patients with ccRCC. We also included 11,367 immune cells from four other individuals derived from the kidney and peripheral blood to facilitate the identification and assessment of ccRCC-specific differences. There is an overall increase in CD8+ T-cell and macrophage populations in tumor-infiltrated immune cells compared to normal renal tissue. We further demonstrate the divergent cell transcriptional states for tumor-infiltrating CD8+ T cells and identify a MKI67 + proliferative subpopulation being a potential culprit for the progression of ccRCC. Using the SCRS gene expression, we found preferential prediction of clinical outcomes and pathological diseases by subcluster assignment. With further characterization and functional validation, our findings may reveal certain subpopulations of immune cells amenable to therapeutic intervention.
Conflict of interest statement
R.W.J. has a financial interest in XSphera Biosciences Inc., a company focused on using ex vivo profiling technology to deliver functional, precision immune-oncology solutions for patients, providers, and drug development companies. R.W.J. interests were reviewed and are managed by Massachusetts General Hospital and Partners HealthCare in accordance with their conflict of interest policies. Y.Z. is on the advisory board of Amgen, Roche Diagnostics, Novartis, Janssen, Eisai, Exelixis, Castle Bioscience, Array, Bayer, Pfizer, Clovis, and EMD Serono. Y.Z. has received institutional clinical trial support from NewLink Genetics, Pfizer, Exelixis, and Eisai. These associations are not related to the work herein described in the paper. Other authors declare no competing interests.
Figures
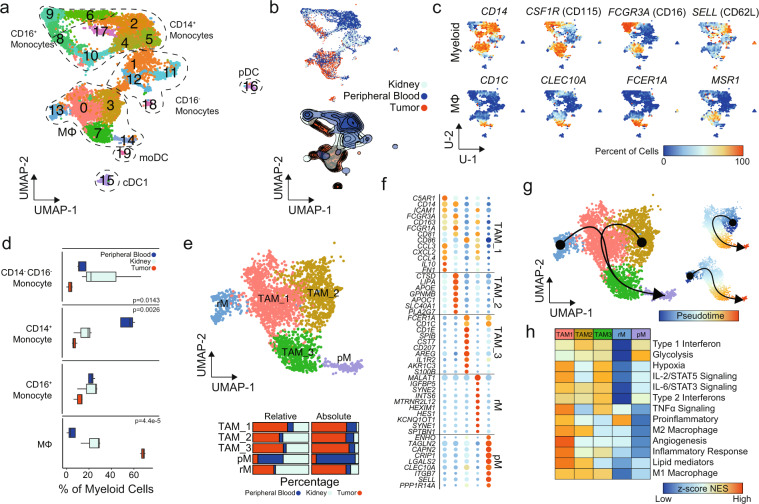
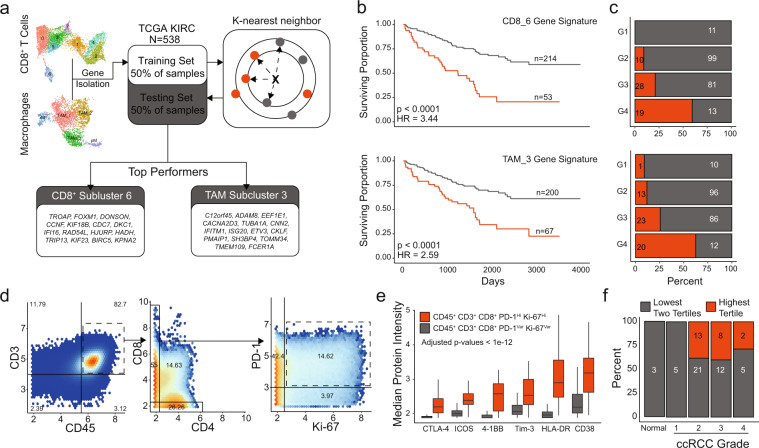
References
-
- Dudani S, et al. First-line (1L) immuno-oncology (IO) combination therapies in metastatic renal cell carcinoma (mRCC): preliminary results from the International Metastatic Renal Cell Carcinoma Database Consortium (IMDC) J. Clin. Oncol. 2019;37:584–584. doi: 10.1200/JCO.2019.37.7_suppl.584. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
