

Integrated spatial genomics reveals global architecture of single nuclei
- PMID: 33505024
- PMCID: PMC7878433
- DOI: 10.1038/s41586-020-03126-2
Integrated spatial genomics reveals global architecture of single nuclei
Abstract
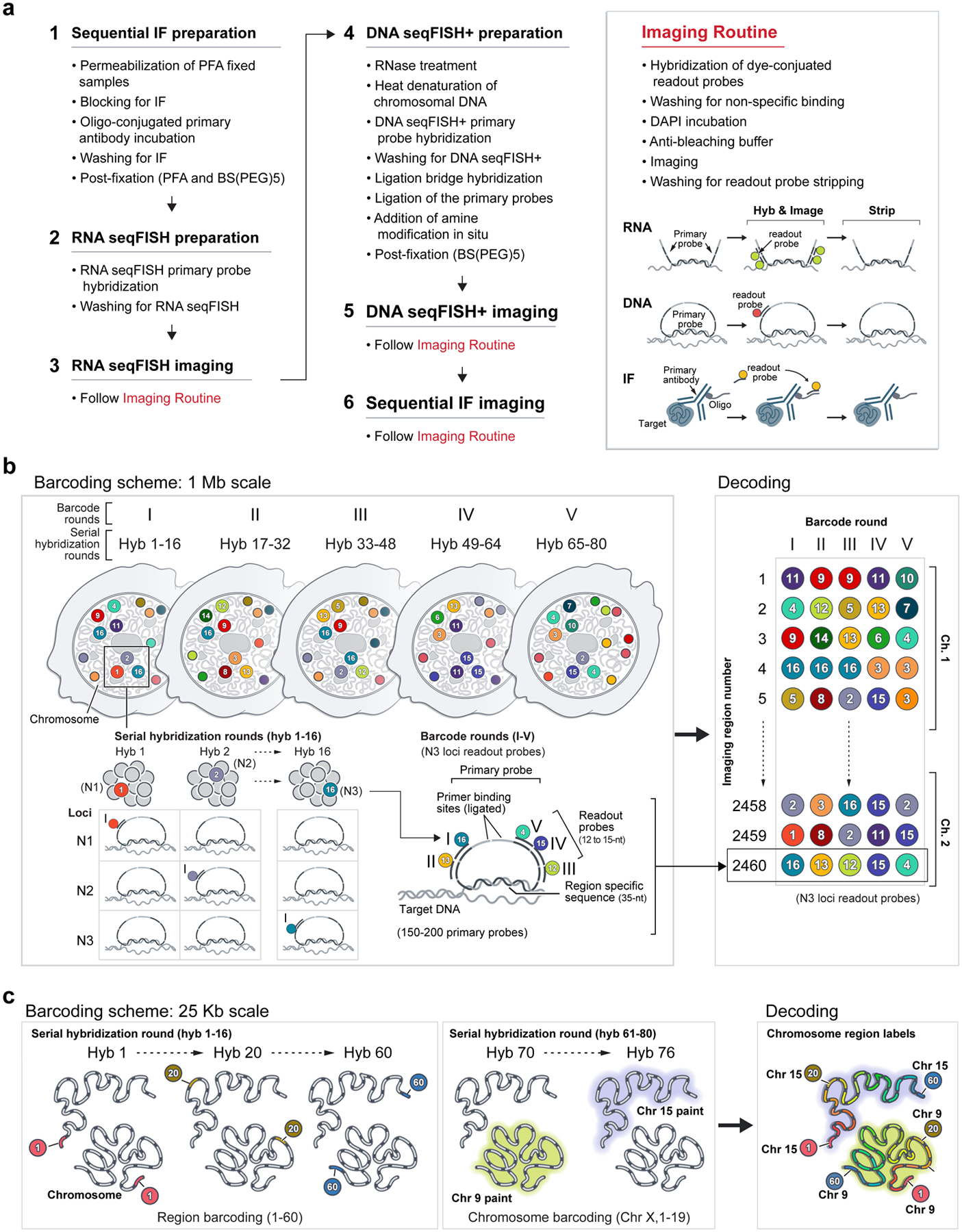
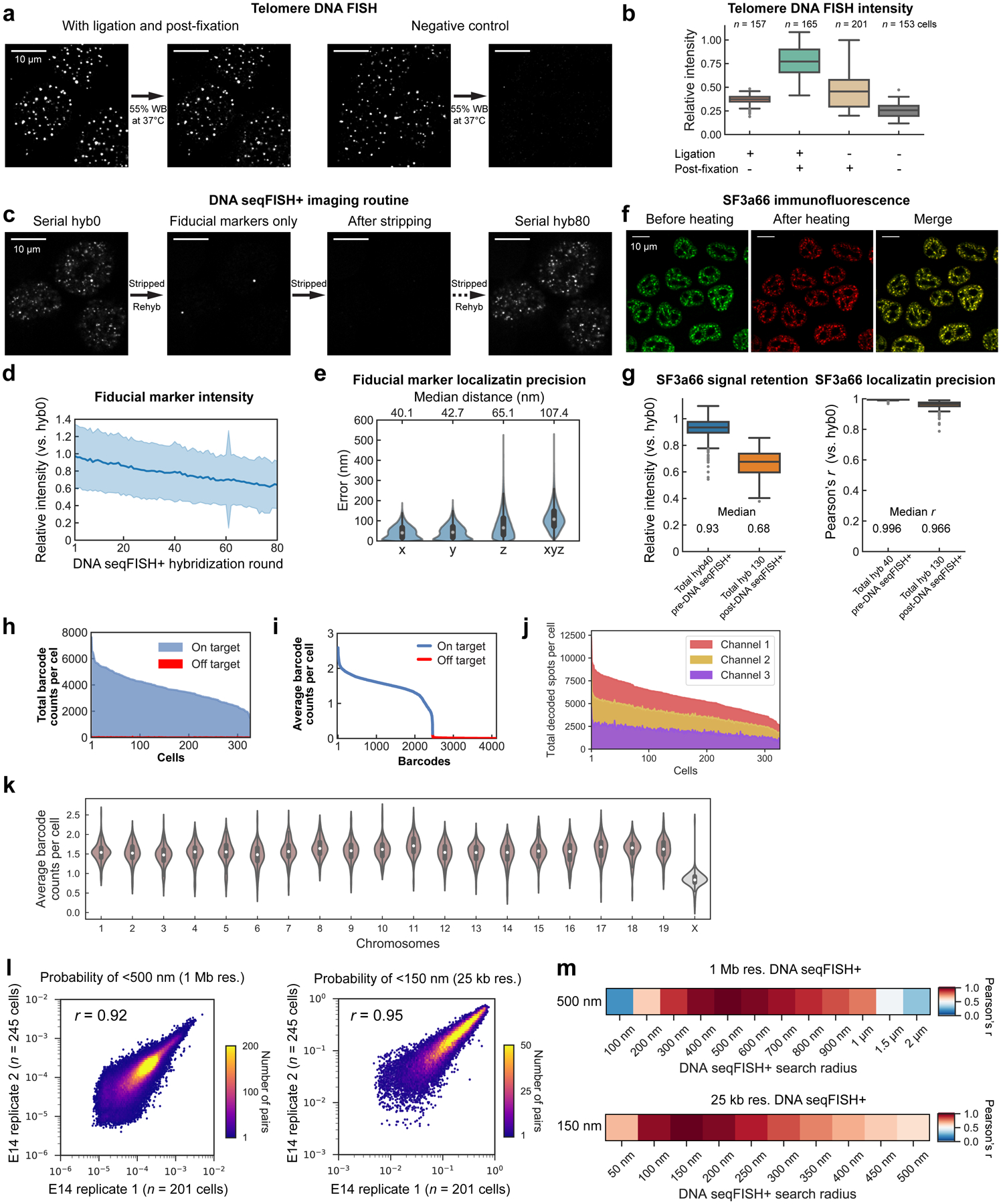
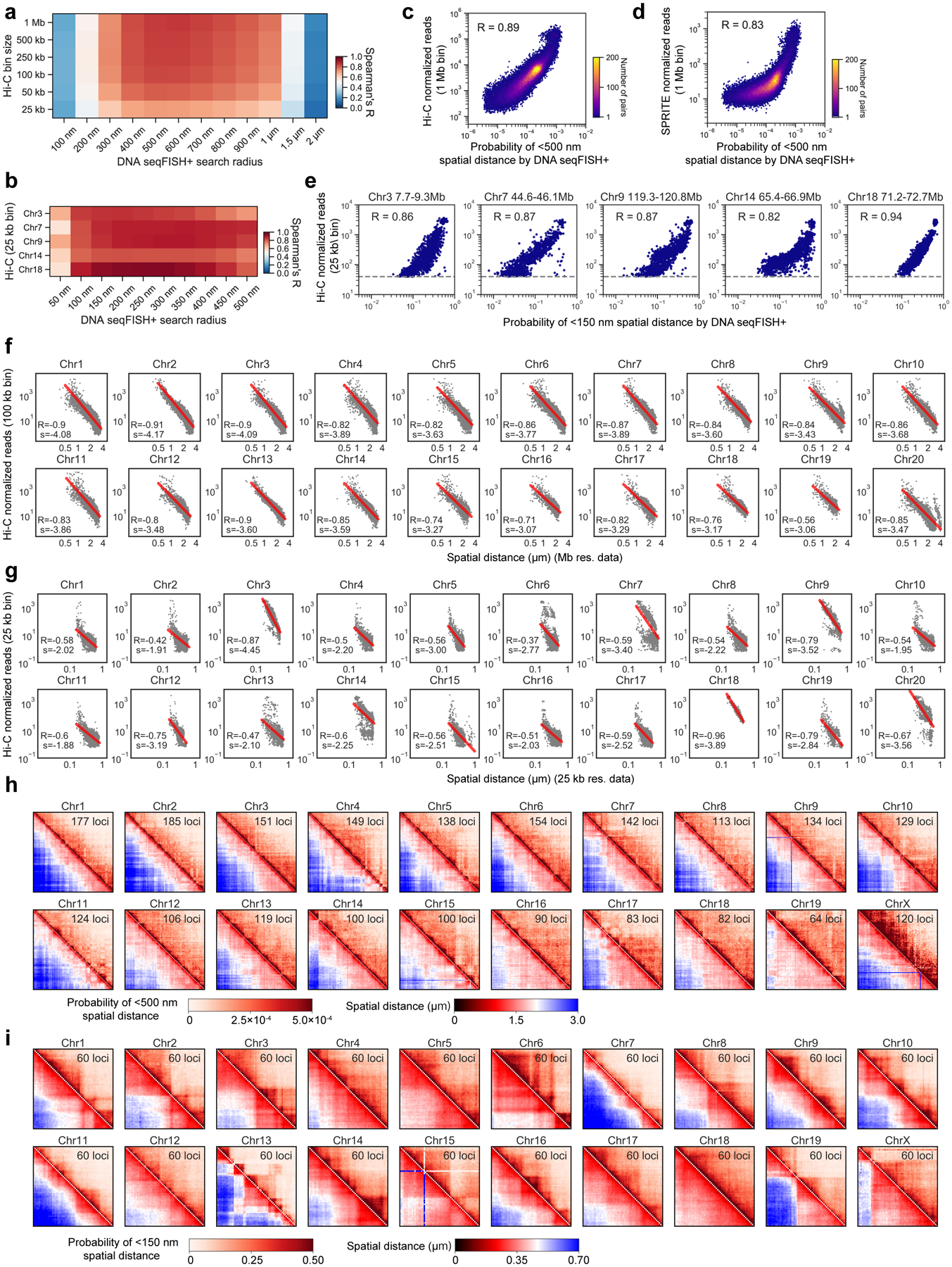
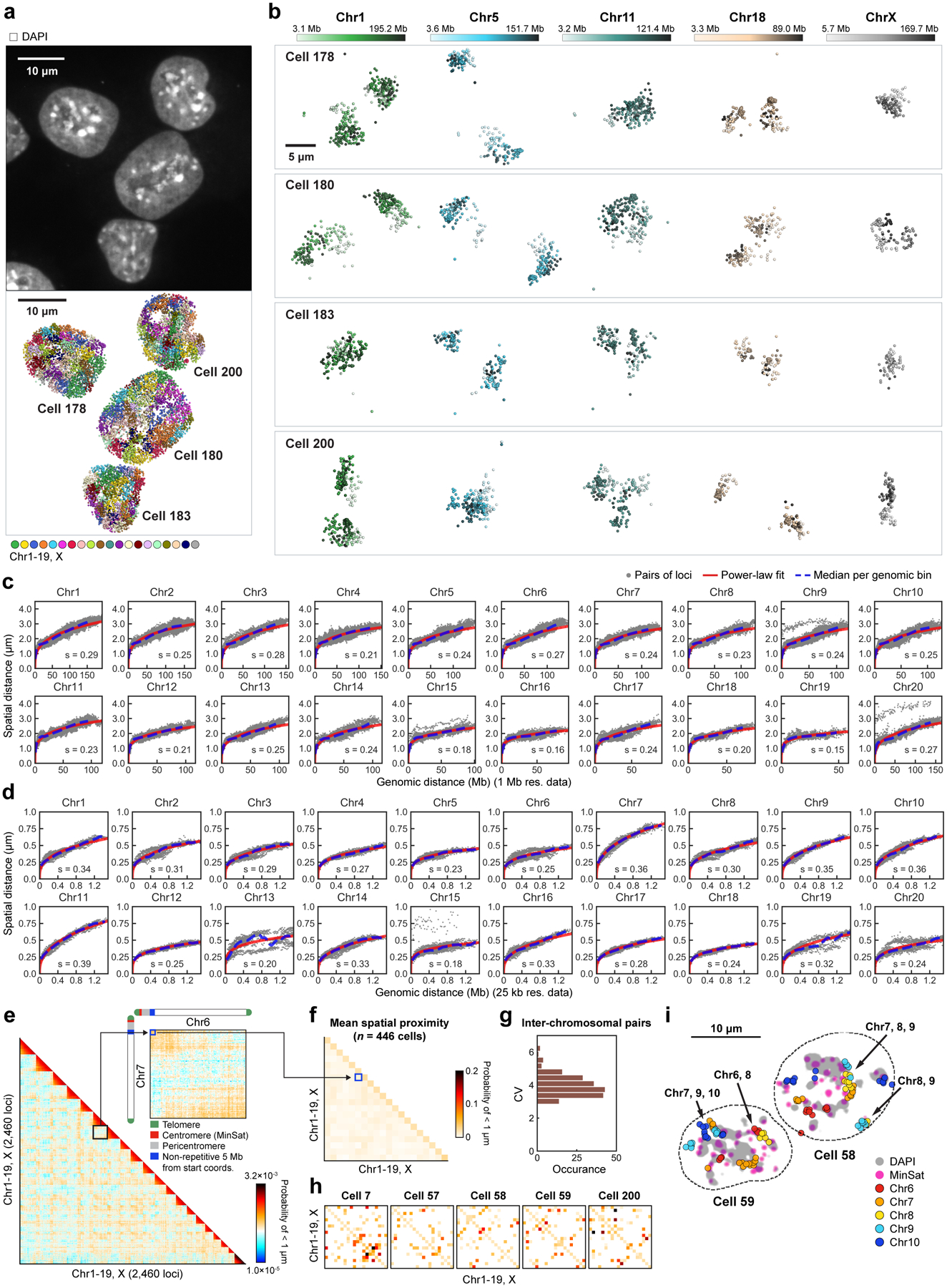
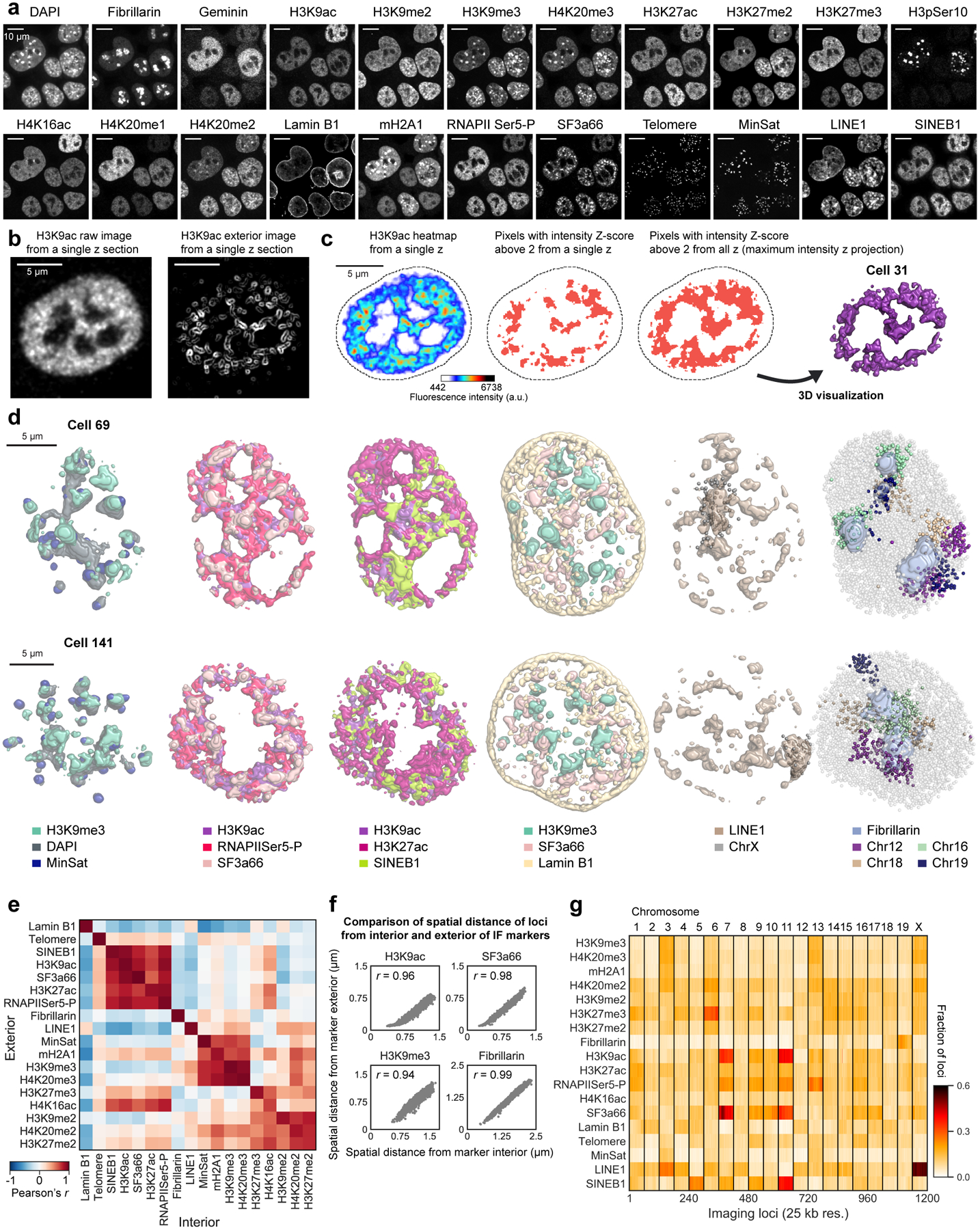
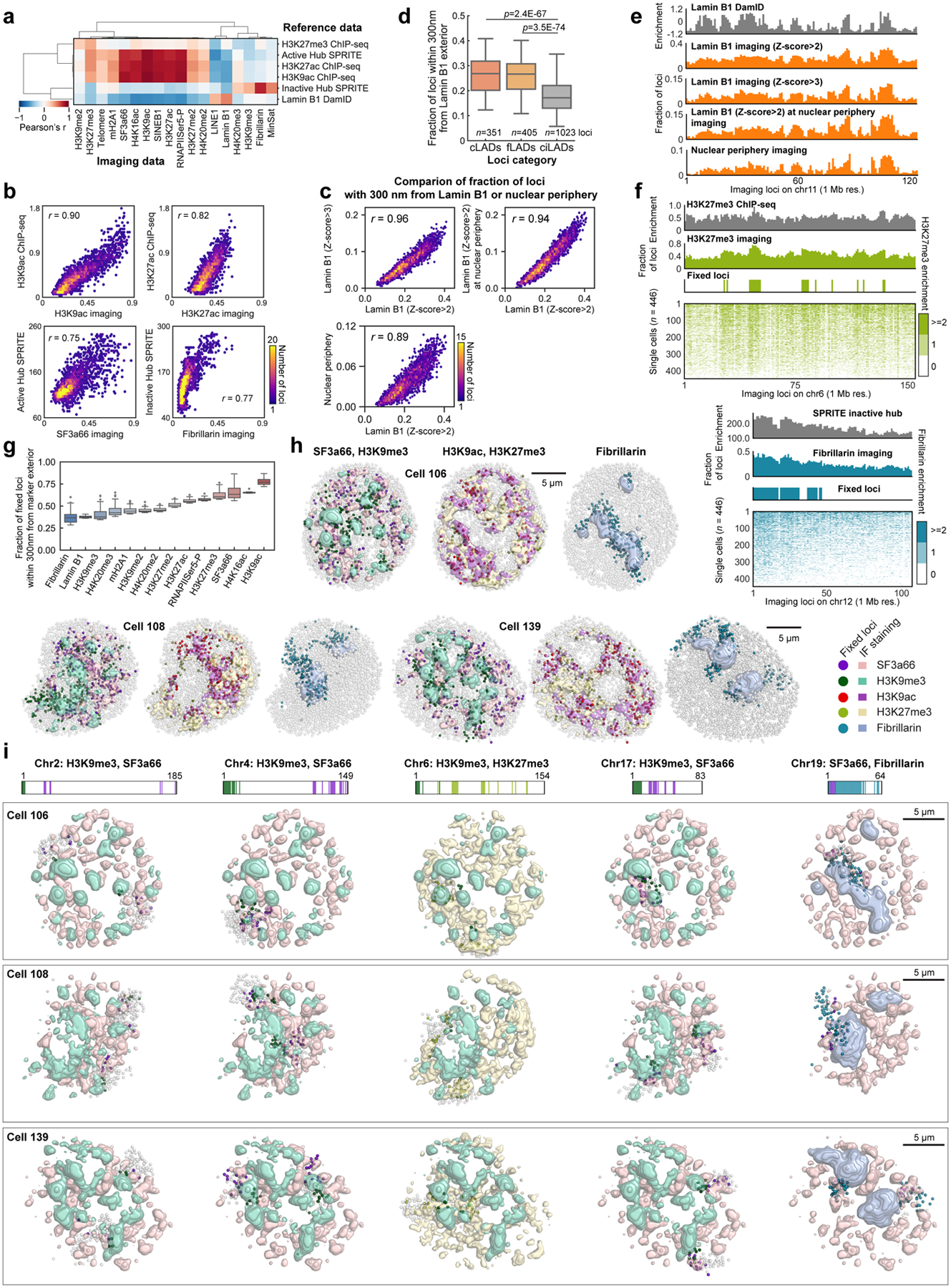
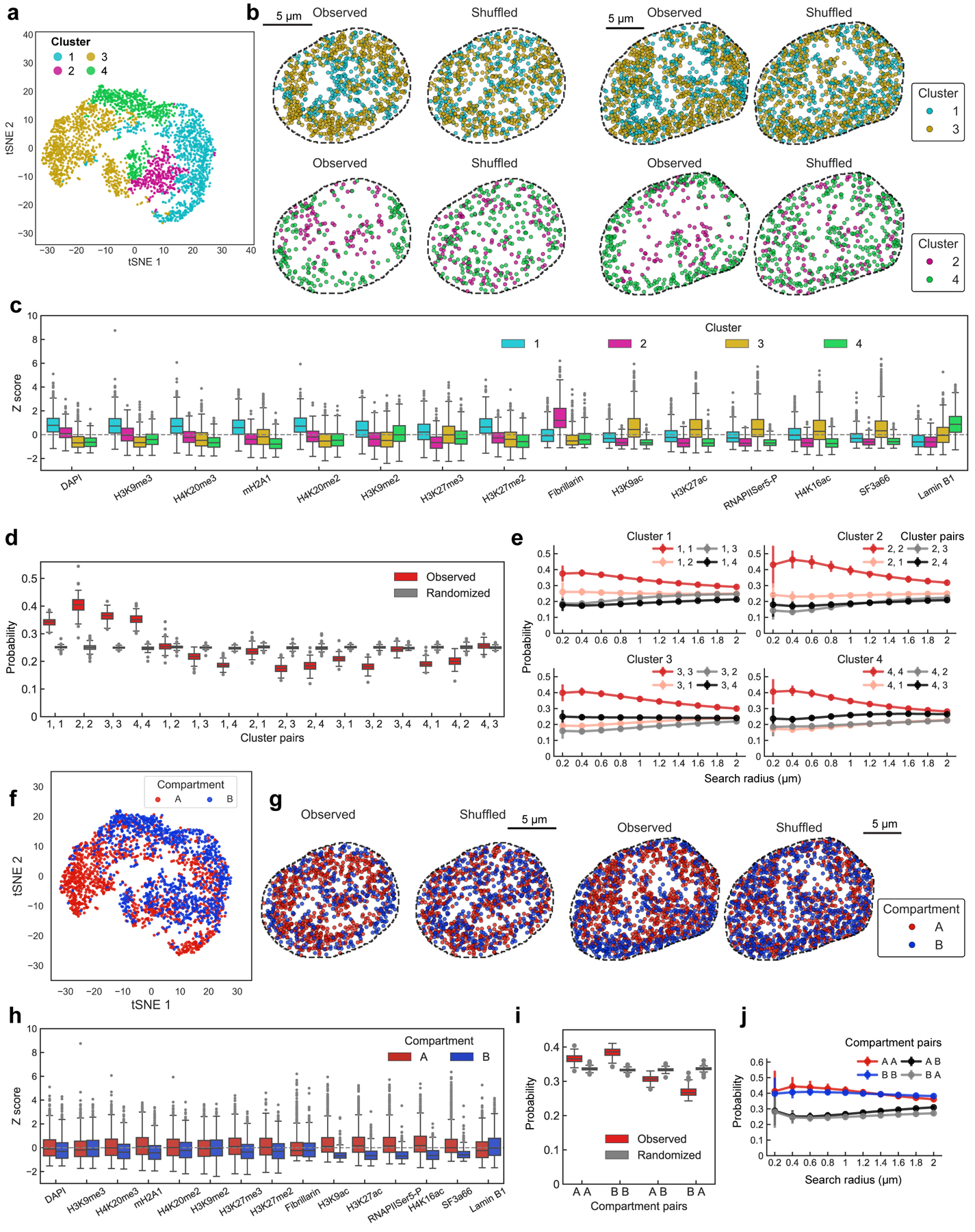
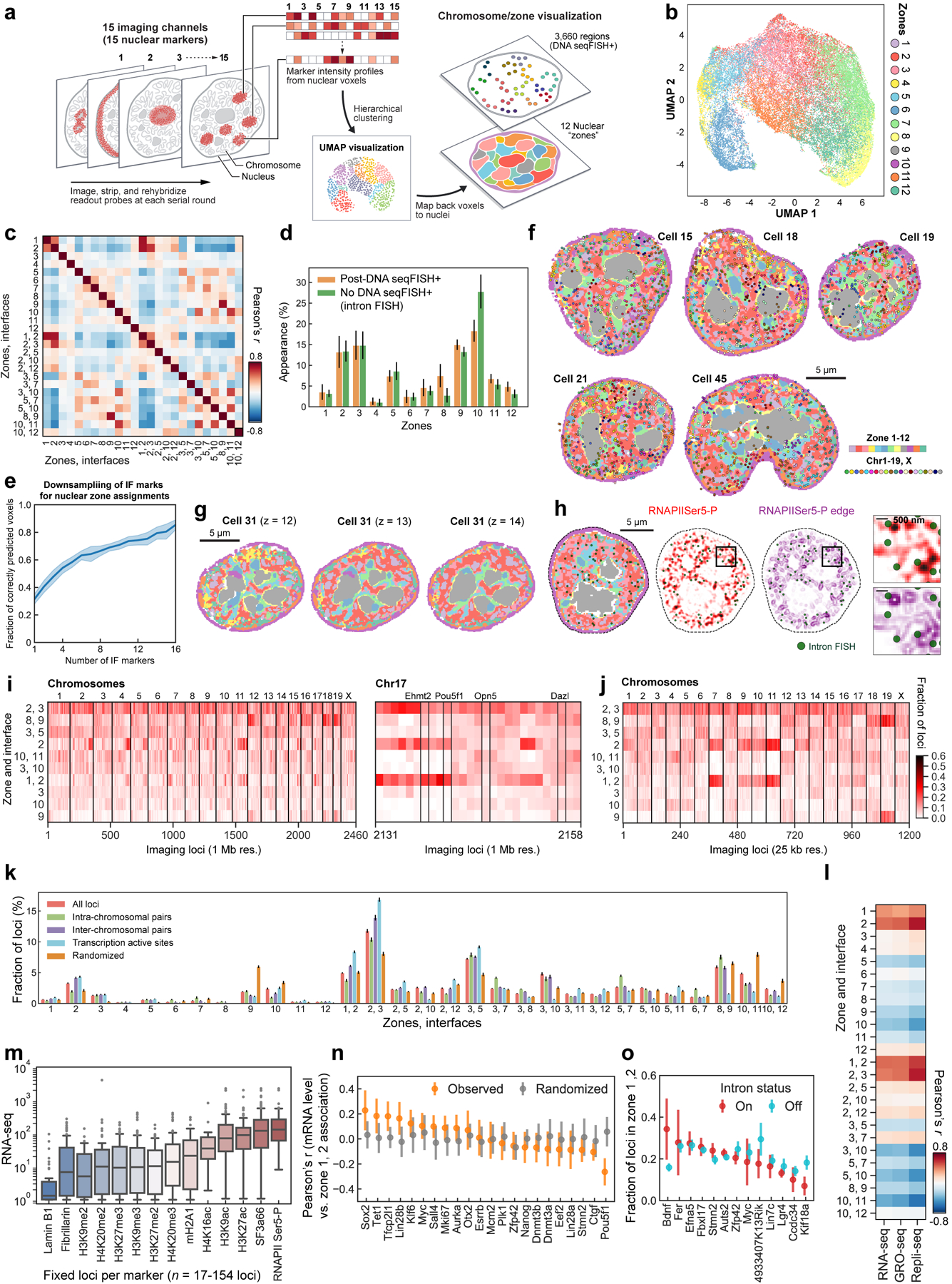
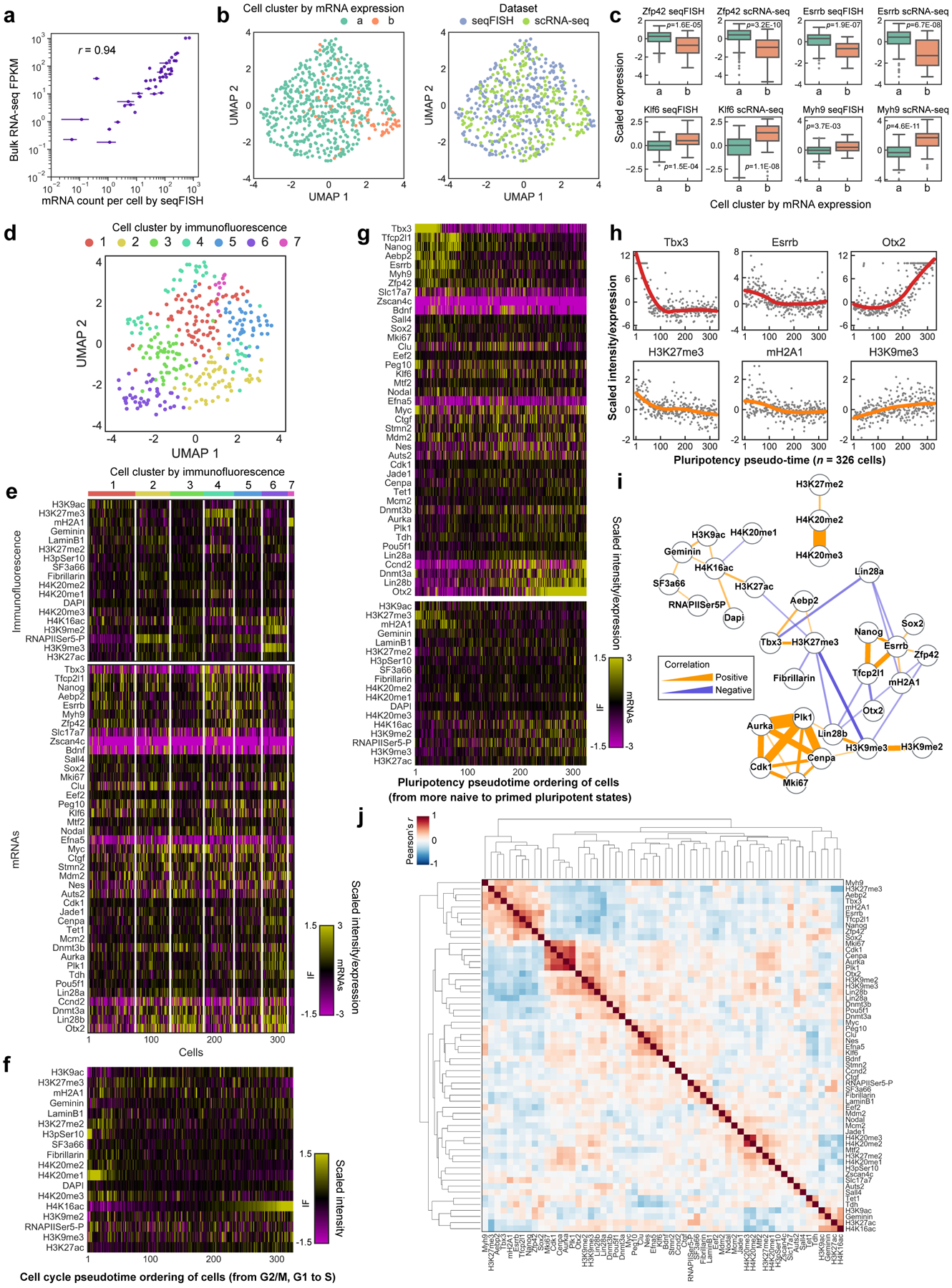
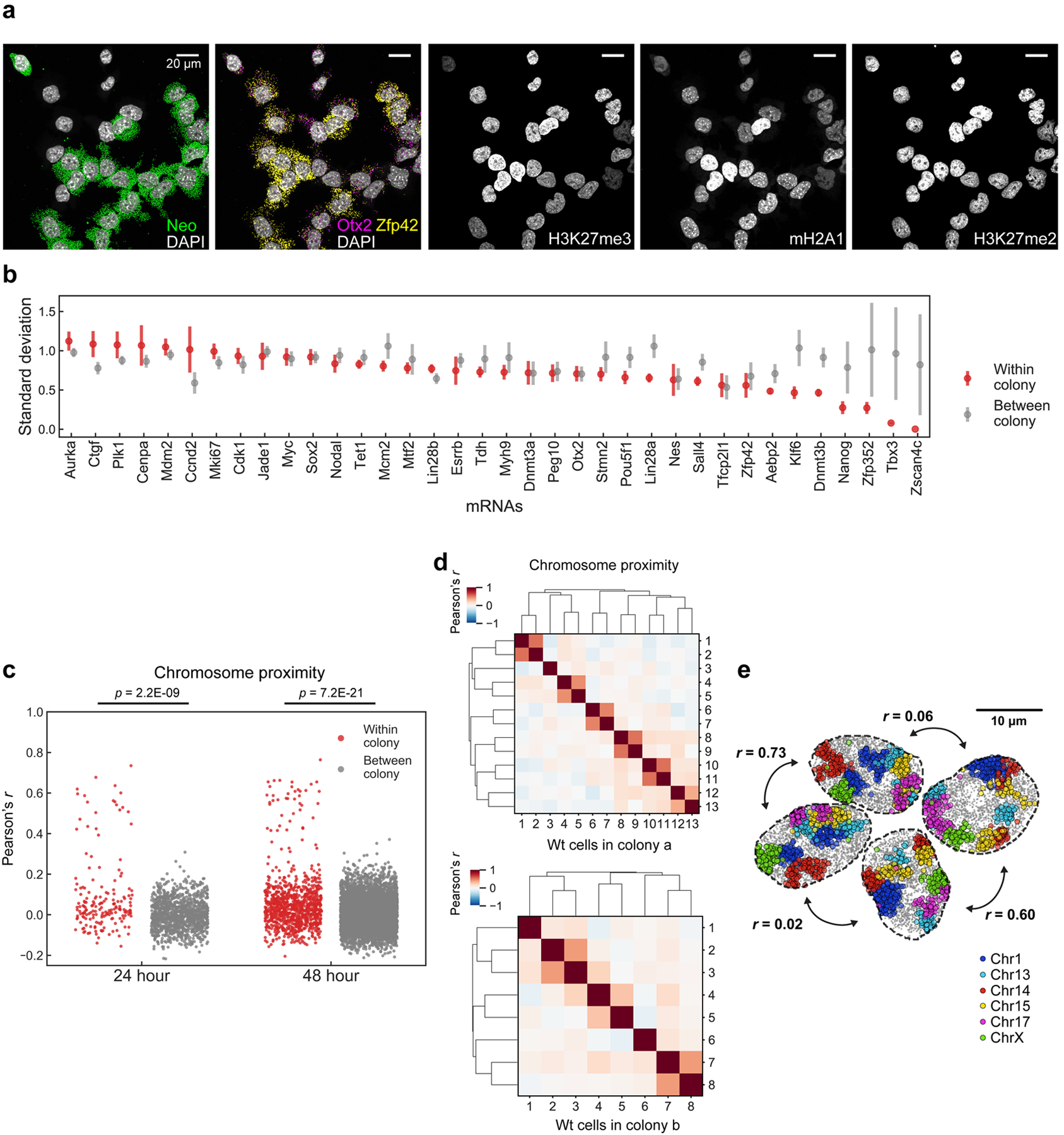
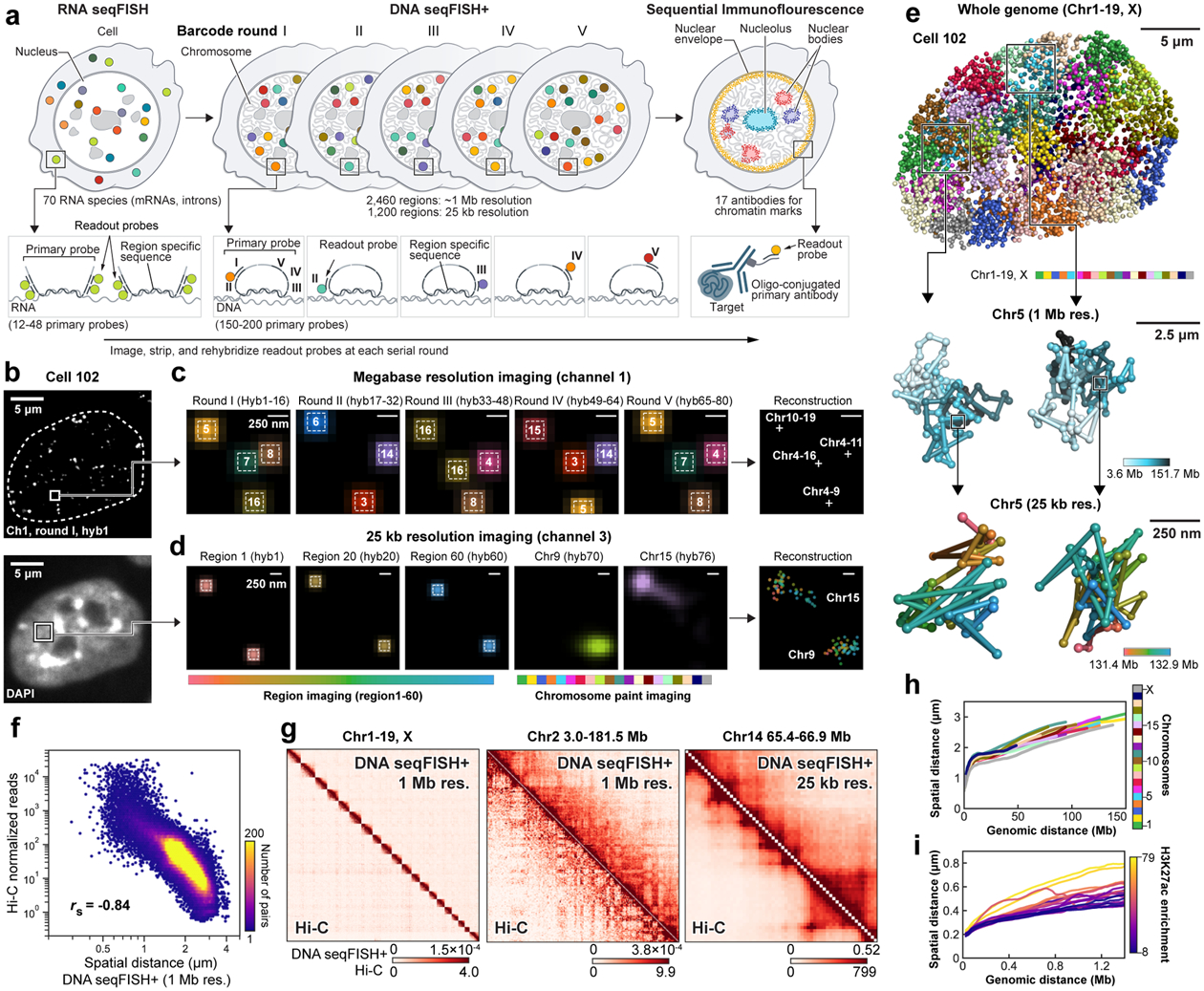
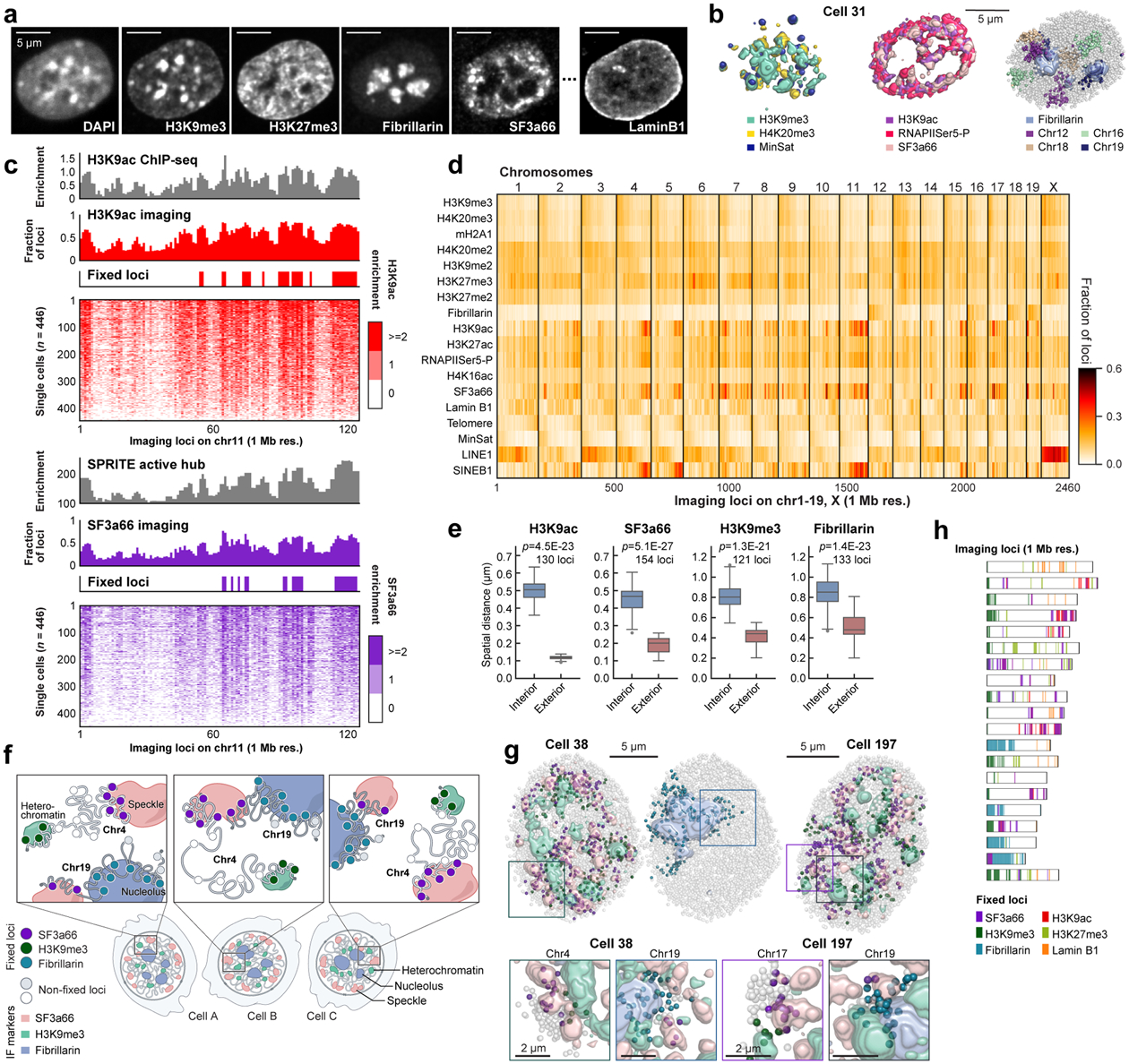
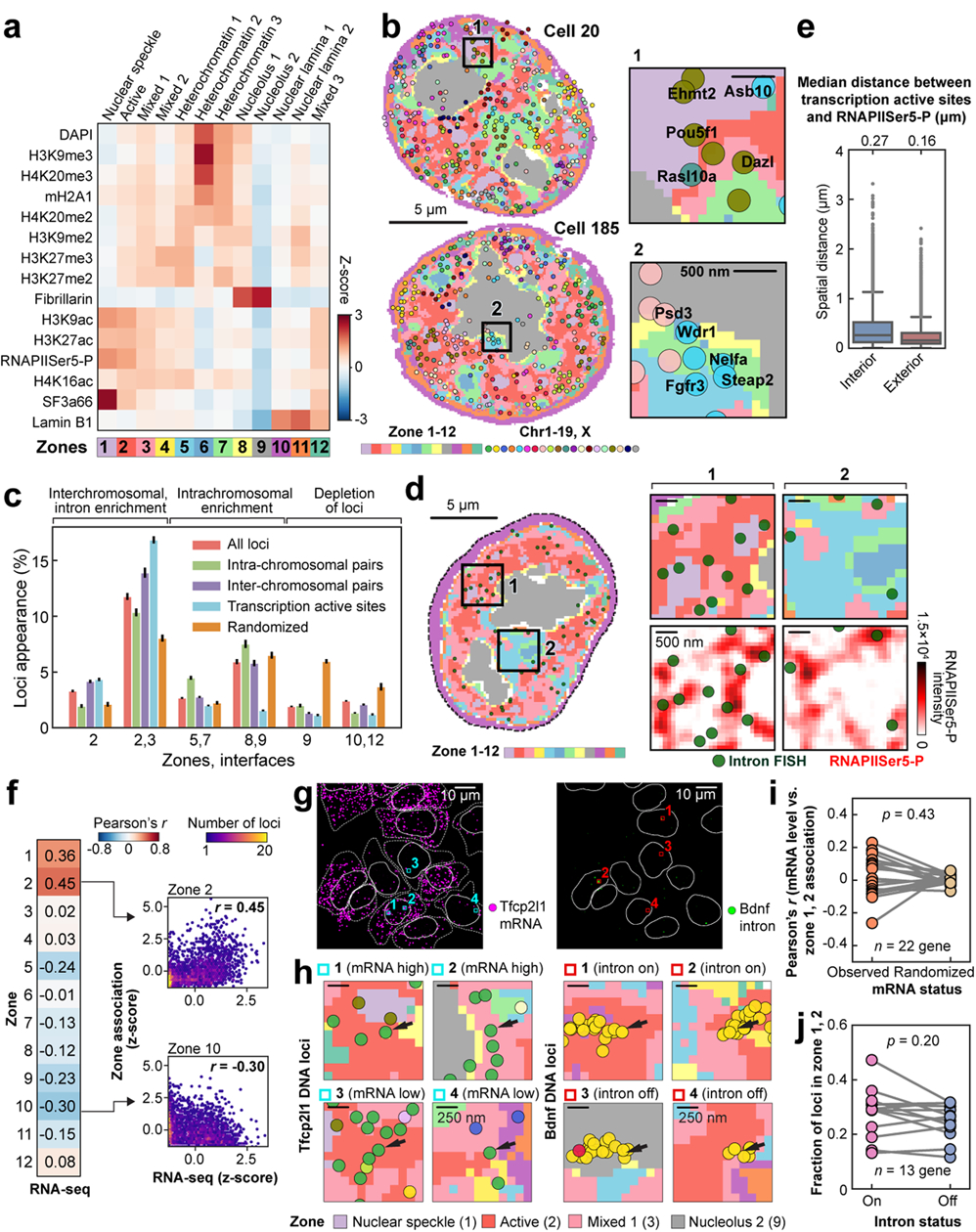
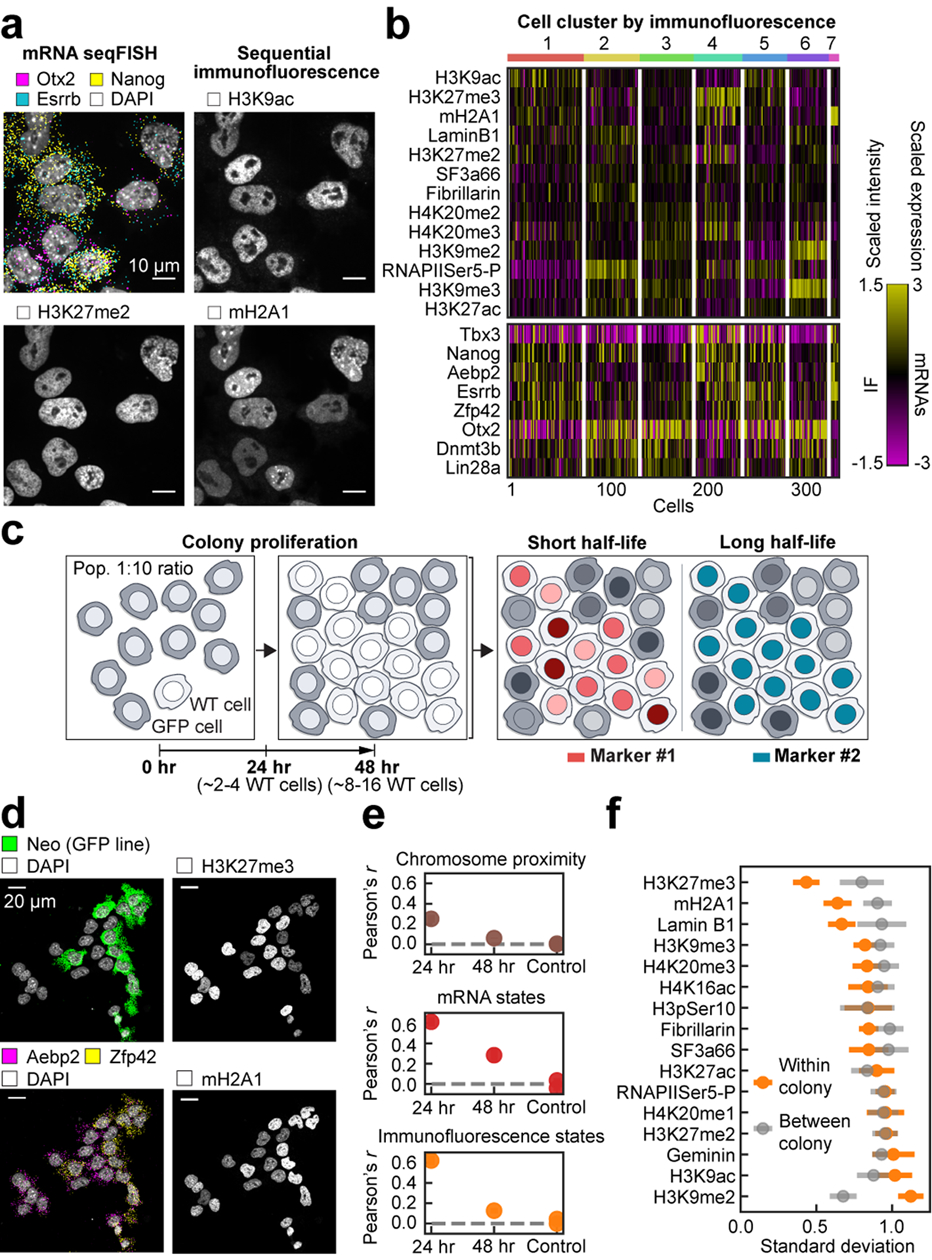
Identifying the relationships between chromosome structures, nuclear bodies, chromatin states and gene expression is an overarching goal of nuclear-organization studies1-4. Because individual cells appear to be highly variable at all these levels5, it is essential to map different modalities in the same cells. Here we report the imaging of 3,660 chromosomal loci in single mouse embryonic stem (ES) cells using DNA seqFISH+, along with 17 chromatin marks and subnuclear structures by sequential immunofluorescence and the expression profile of 70 RNAs. Many loci were invariably associated with immunofluorescence marks in single mouse ES cells. These loci form 'fixed points' in the nuclear organizations of single cells and often appear on the surfaces of nuclear bodies and zones defined by combinatorial chromatin marks. Furthermore, highly expressed genes appear to be pre-positioned to active nuclear zones, independent of bursting dynamics in single cells. Our analysis also uncovered several distinct mouse ES cell subpopulations with characteristic combinatorial chromatin states. Using clonal analysis, we show that the global levels of some chromatin marks, such as H3 trimethylation at lysine 27 (H3K27me3) and macroH2A1 (mH2A1), are heritable over at least 3-4 generations, whereas other marks fluctuate on a faster time scale. This seqFISH+-based spatial multimodal approach can be used to explore nuclear organization and cell states in diverse biological systems.
Conflict of interest statement
Competing Interest:
L.C. is a co-founder of Spatial Genomics Inc.
Figures
Comment in
-
Grand designs of the nucleus.Nat Rev Genet. 2021 Apr;22(4):200-201. doi: 10.1038/s41576-021-00336-w. Nat Rev Genet. 2021. PMID: 33608687 No abstract available.
References
-
- Kelsey G, Stegle O & Reik W Single-cell epigenomics: Recording the past and predicting the future. Science vol. 358 69–75 (2017). - PubMed
-
- Kempfer R & Pombo A Methods for mapping 3D chromosome architecture. Nat. Rev. Genet 21, 207–226 (2020). - PubMed
-
- Zhu C, Preissl S & Ren B Single-cell multimodal omics: the power of many. Nat. Methods 17, 11–14 (2020). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
