

Systematic analysis of binding of transcription factors to noncoding variants
- PMID: 33505025
- PMCID: PMC9367673
- DOI: 10.1038/s41586-021-03211-0
Systematic analysis of binding of transcription factors to noncoding variants
Abstract
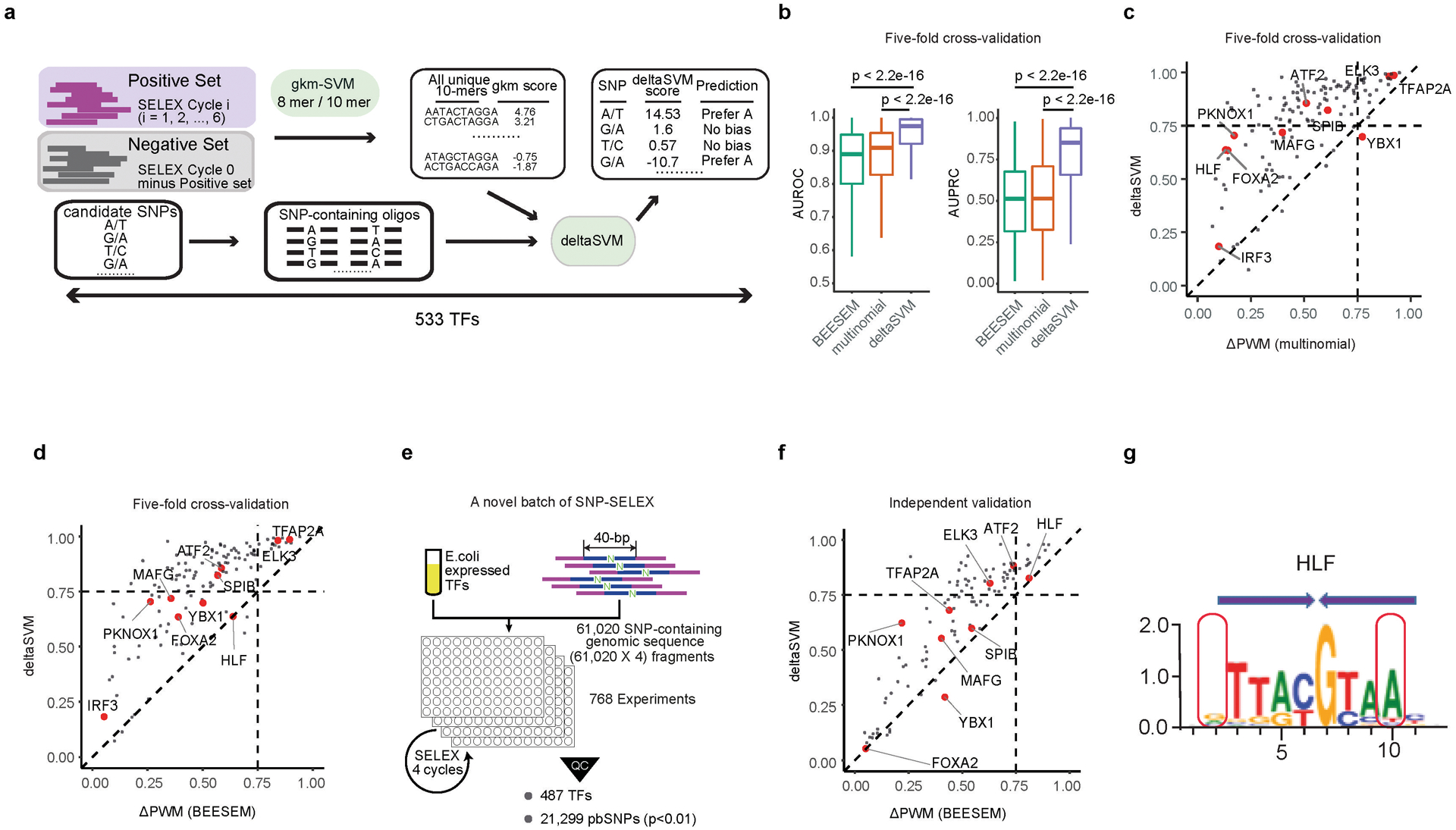
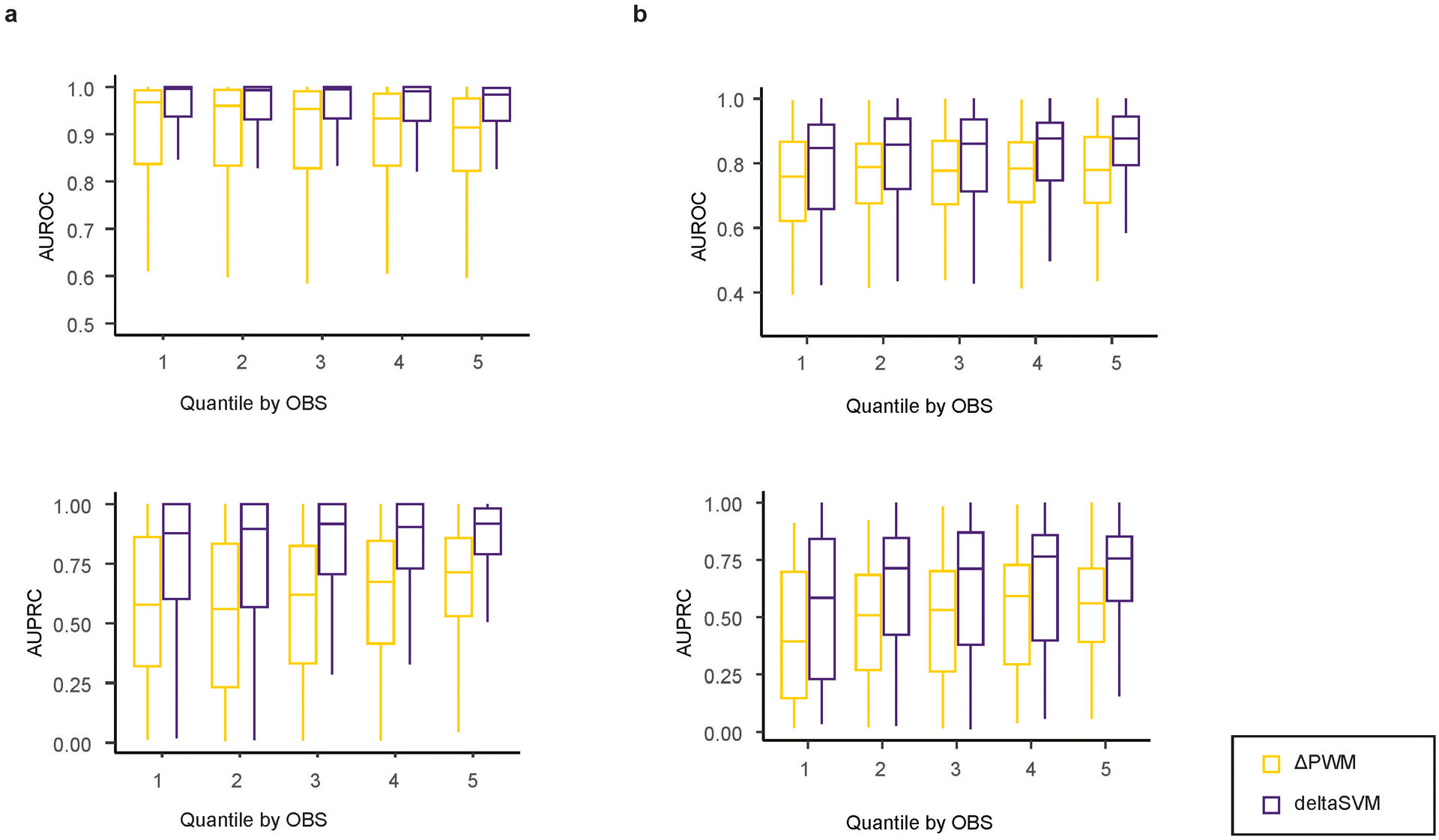
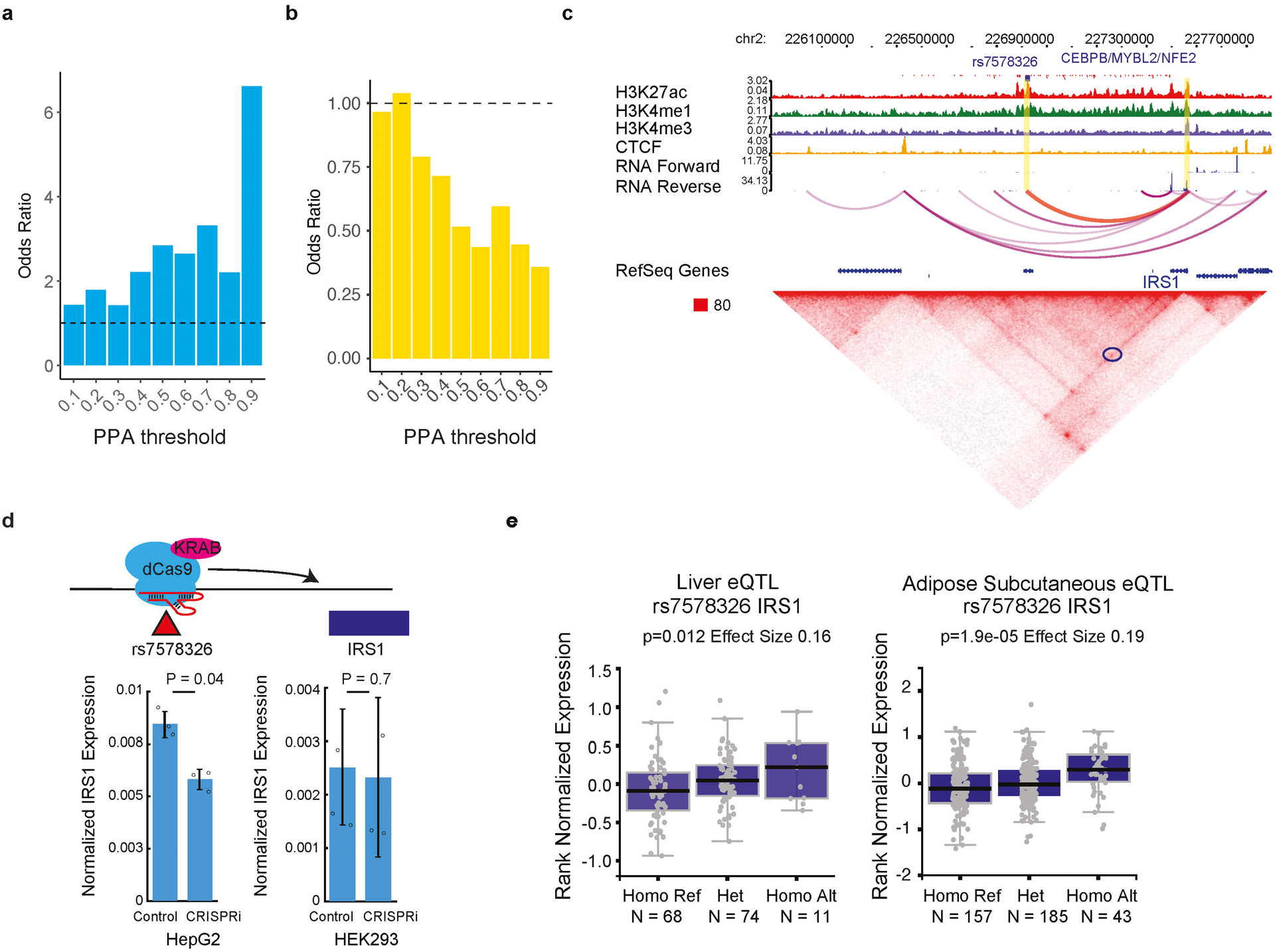
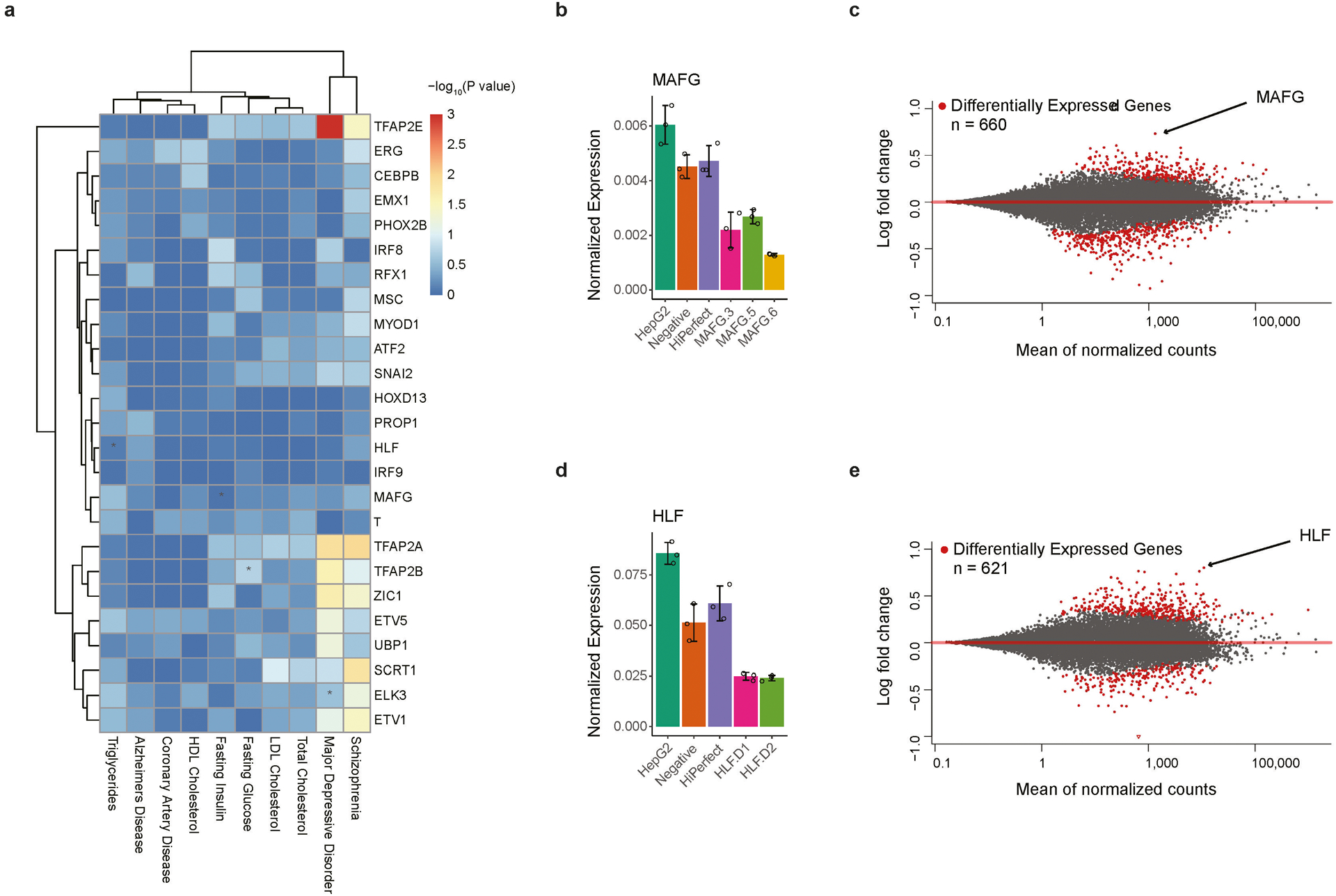
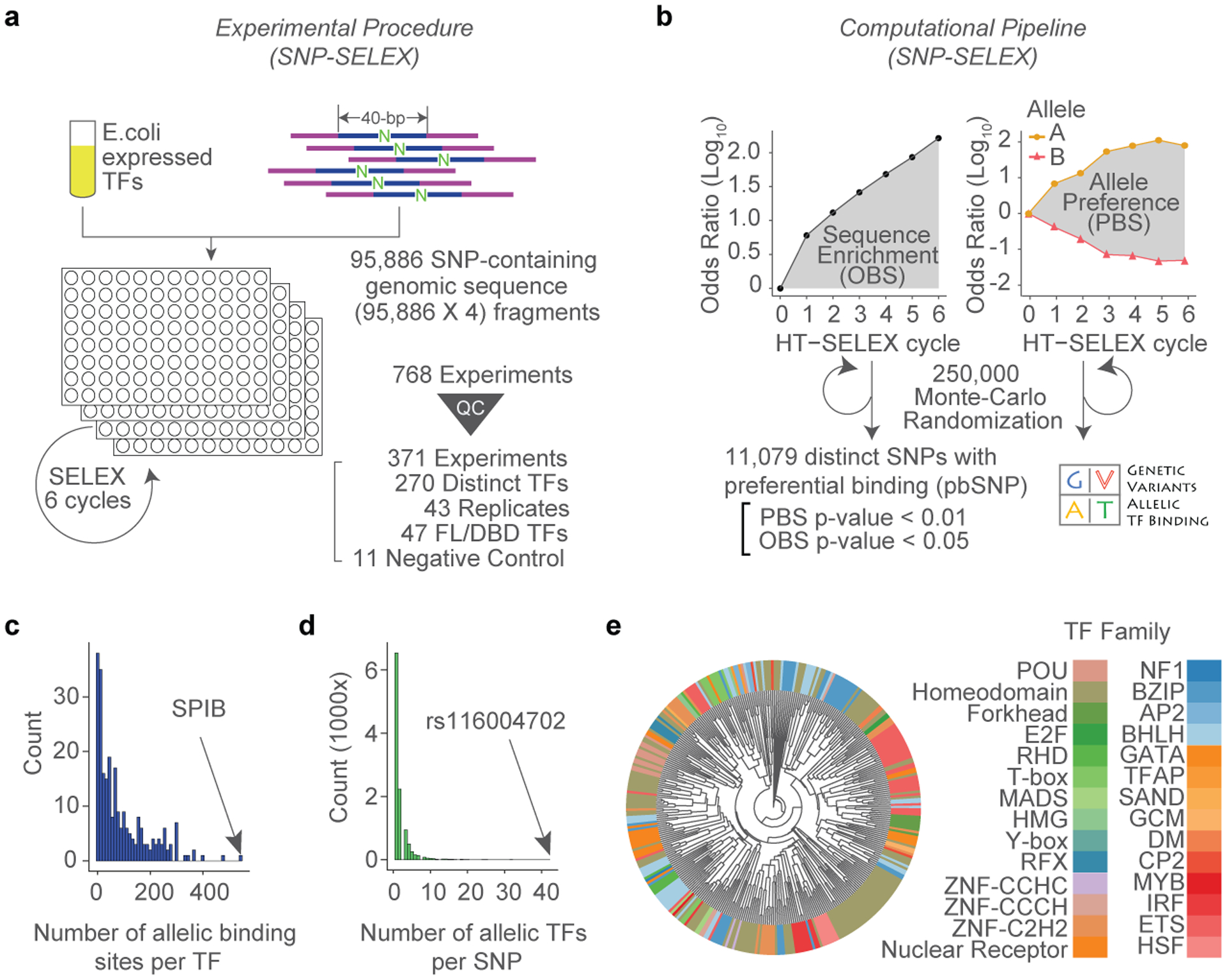
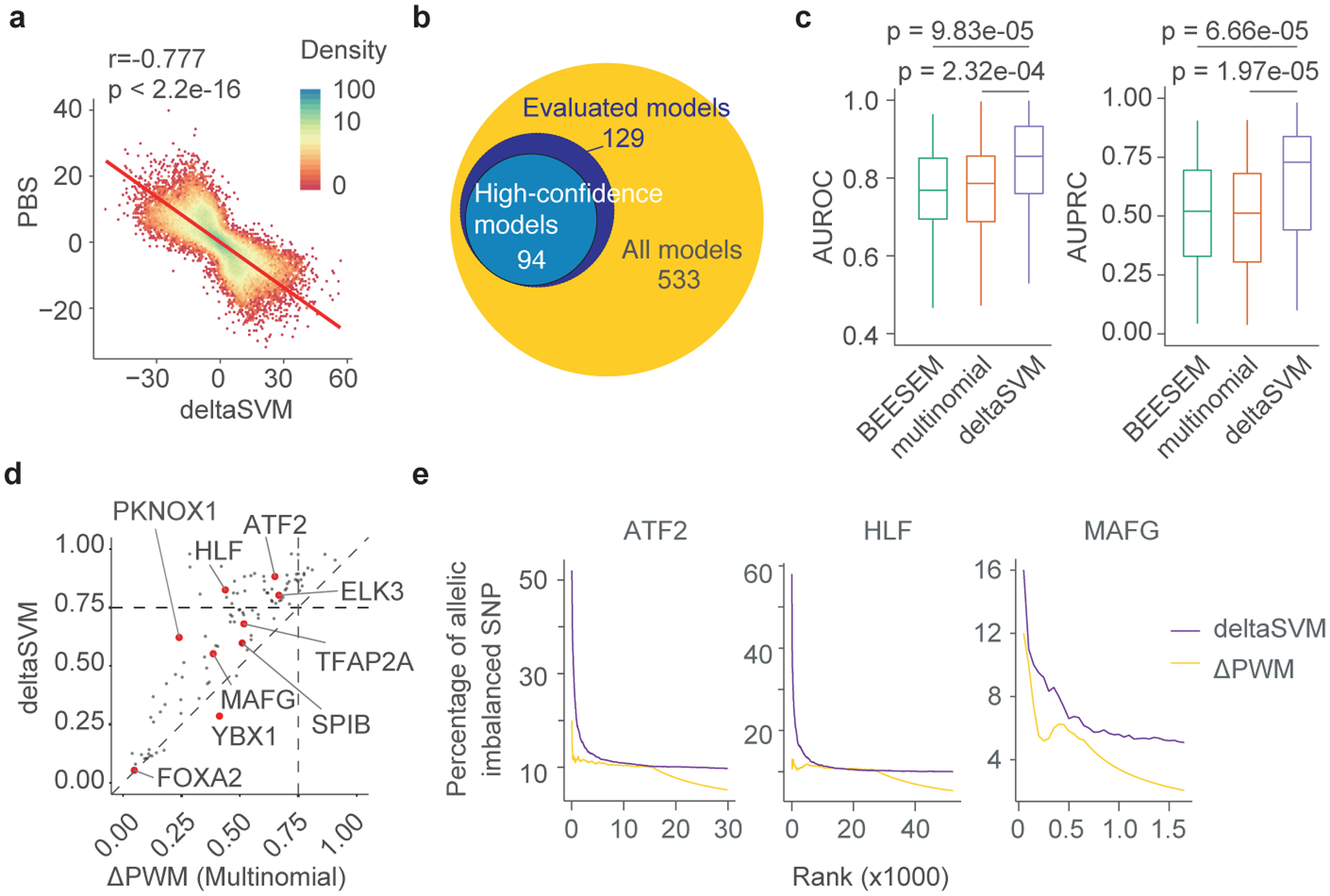
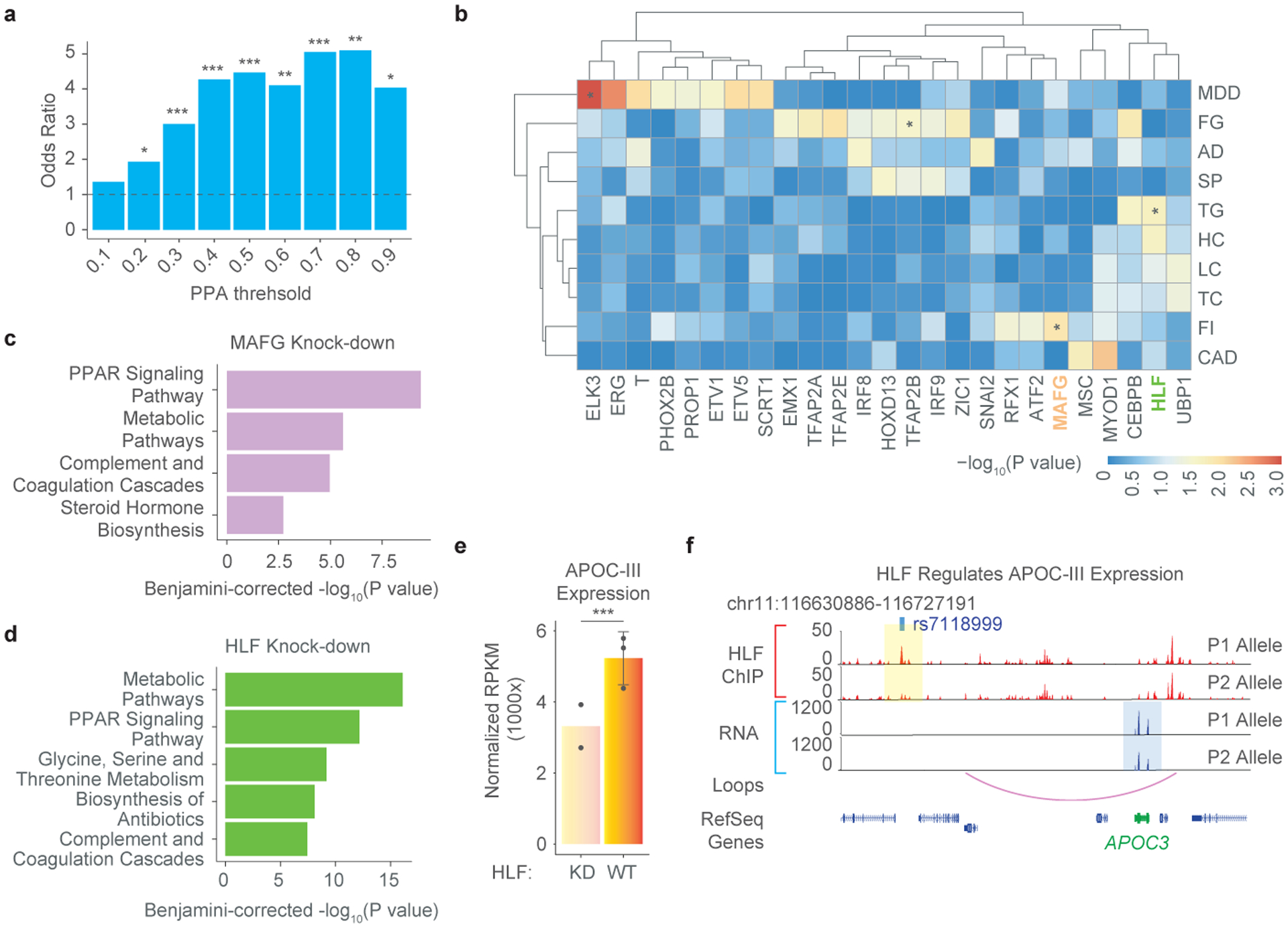
Many sequence variants have been linked to complex human traits and diseases1, but deciphering their biological functions remains challenging, as most of them reside in noncoding DNA. Here we have systematically assessed the binding of 270 human transcription factors to 95,886 noncoding variants in the human genome using an ultra-high-throughput multiplex protein-DNA binding assay, termed single-nucleotide polymorphism evaluation by systematic evolution of ligands by exponential enrichment (SNP-SELEX). The resulting 828 million measurements of transcription factor-DNA interactions enable estimation of the relative affinity of these transcription factors to each variant in vitro and evaluation of the current methods to predict the effects of noncoding variants on transcription factor binding. We show that the position weight matrices of most transcription factors lack sufficient predictive power, whereas the support vector machine combined with the gapped k-mer representation show much improved performance, when assessed on results from independent SNP-SELEX experiments involving a new set of 61,020 sequence variants. We report highly predictive models for 94 human transcription factors and demonstrate their utility in genome-wide association studies and understanding of the molecular pathways involved in diverse human traits and diseases.
Figures
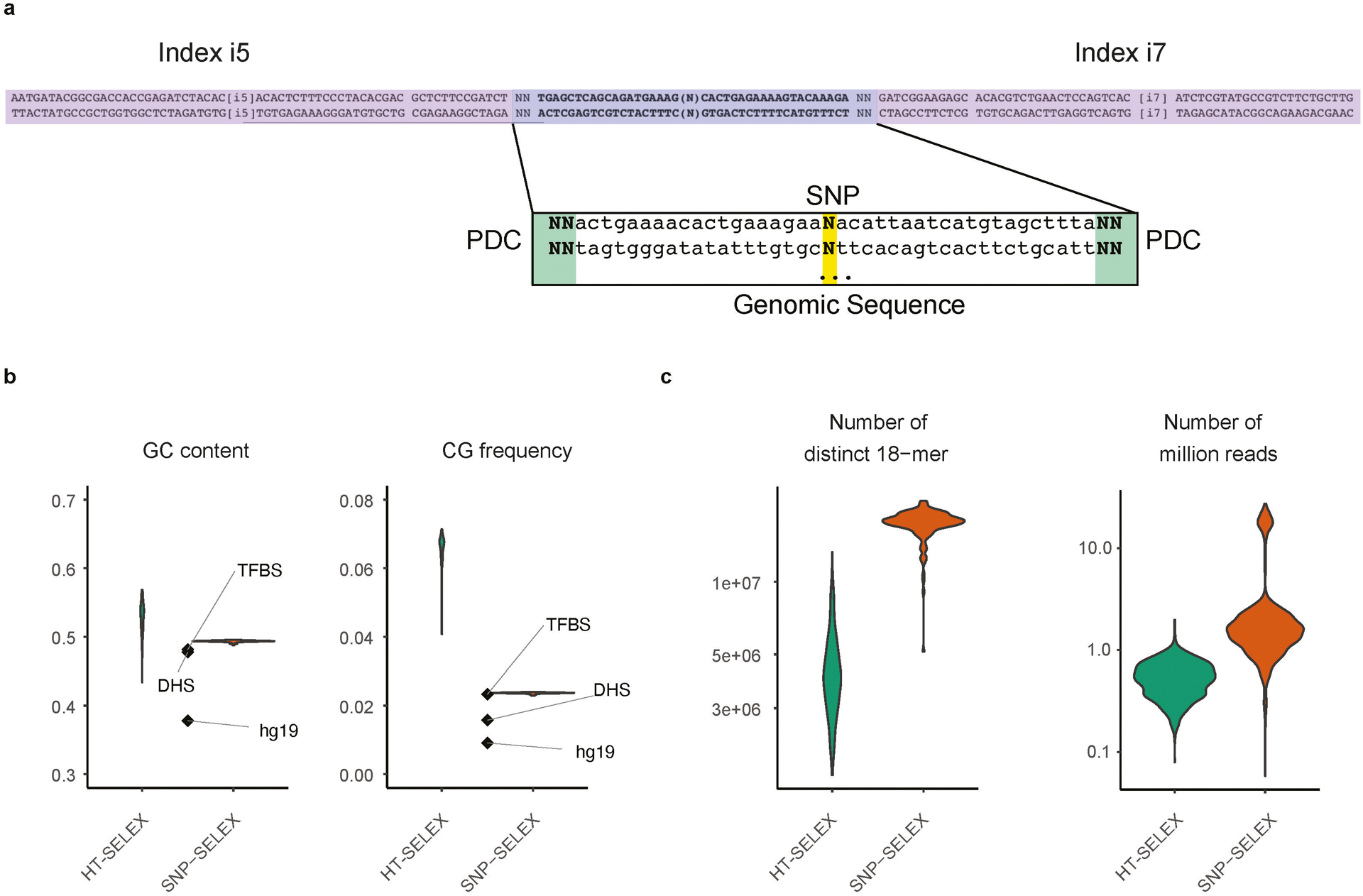
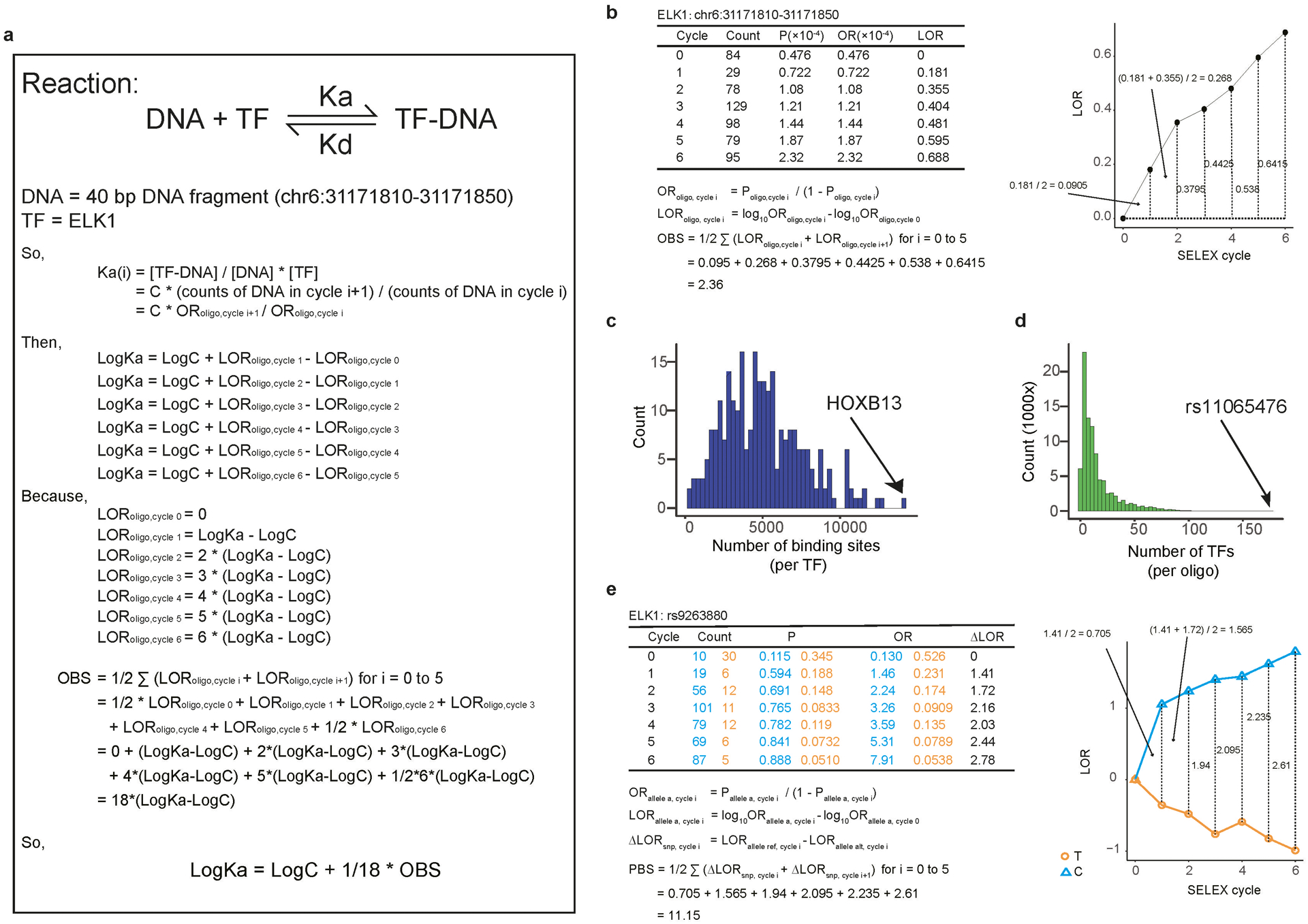

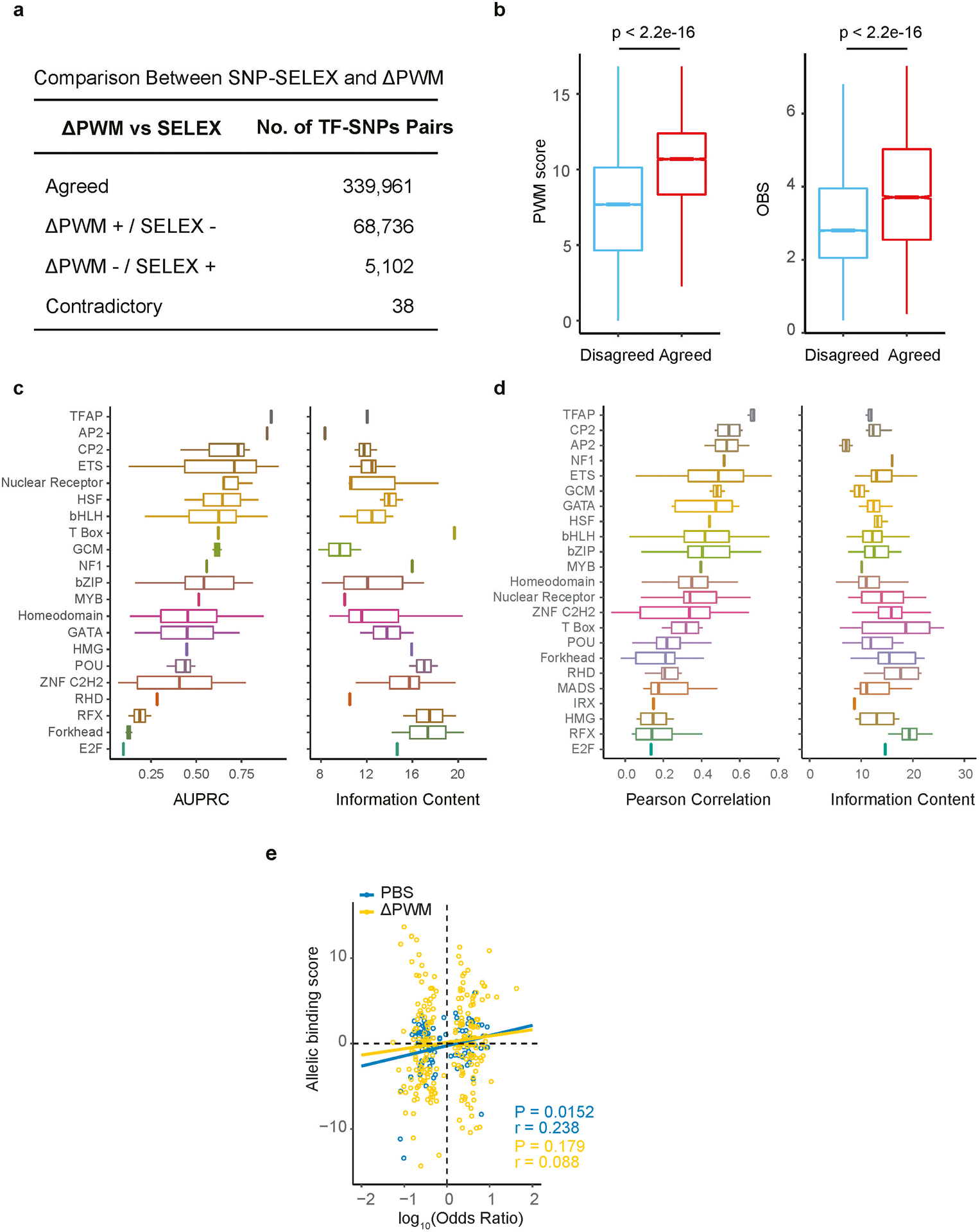
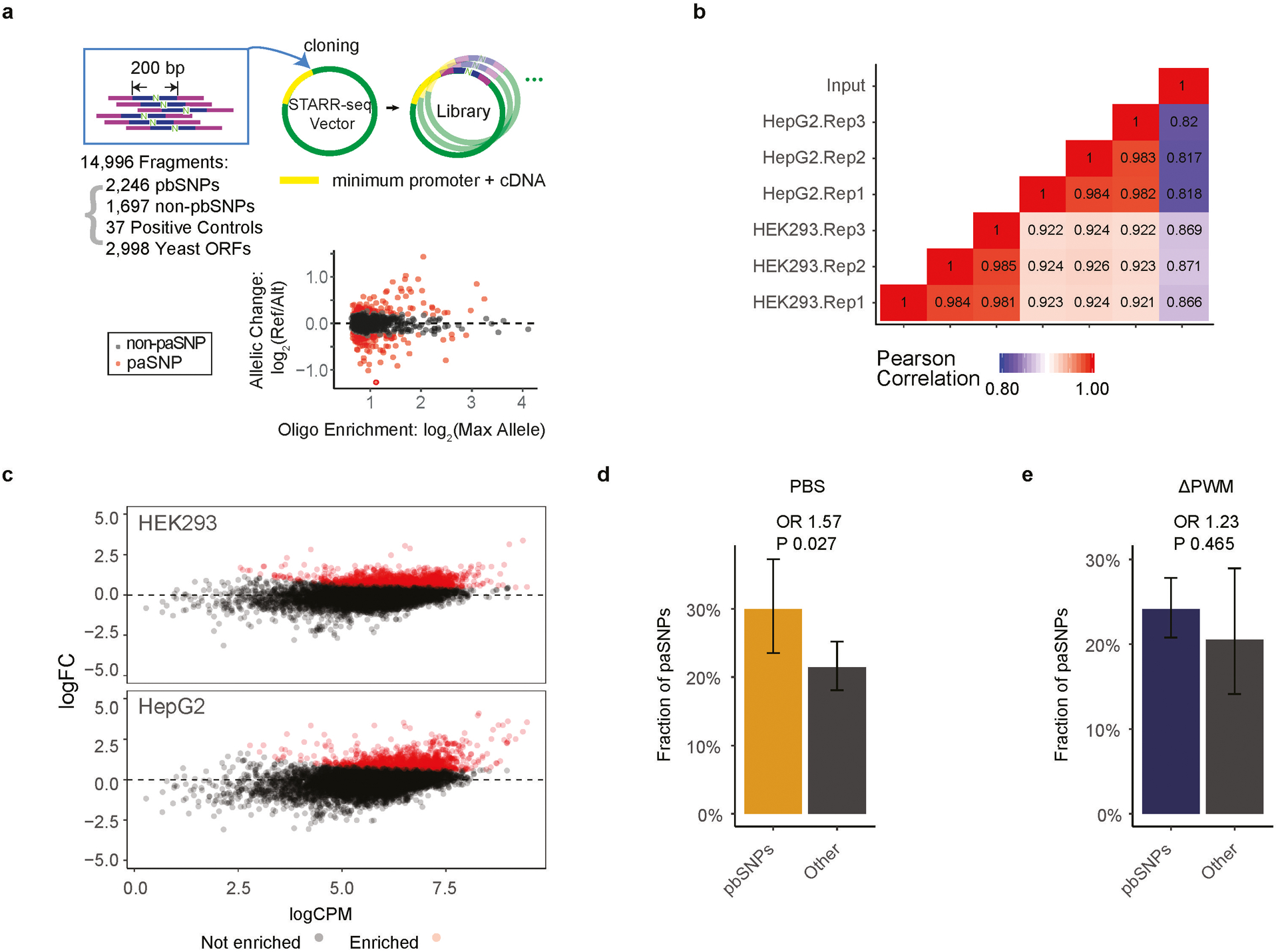


Similar articles
-
SELEX (Systematic Evolution of Ligands by EXponential Enrichment), as a powerful tool for deciphering the protein-DNA interaction space.Methods Mol Biol. 2011;754:249-58. doi: 10.1007/978-1-61779-154-3_14. Methods Mol Biol. 2011. PMID: 21720957
-
SELEX-Seq: A Method to Determine DNA Binding Specificities of Plant Transcription Factors.Methods Mol Biol. 2017;1629:67-82. doi: 10.1007/978-1-4939-7125-1_6. Methods Mol Biol. 2017. PMID: 28623580
-
GERV: a statistical method for generative evaluation of regulatory variants for transcription factor binding.Bioinformatics. 2016 Feb 15;32(4):490-6. doi: 10.1093/bioinformatics/btv565. Epub 2015 Oct 17. Bioinformatics. 2016. PMID: 26476779 Free PMC article.
-
In vitro DNA-binding profile of transcription factors: methods and new insights.J Endocrinol. 2011 Jul;210(1):15-27. doi: 10.1530/JOE-11-0010. Epub 2011 Mar 9. J Endocrinol. 2011. PMID: 21389103 Review.
-
Regulatory SNPs: Altered Transcription Factor Binding Sites Implicated in Complex Traits and Diseases.Int J Mol Sci. 2021 Jun 16;22(12):6454. doi: 10.3390/ijms22126454. Int J Mol Sci. 2021. PMID: 34208629 Free PMC article. Review.
Cited by
-
Multi-Omics Analysis Revealed the rSNPs Potentially Involved in T2DM Pathogenic Mechanism and Metformin Response.Int J Mol Sci. 2024 Aug 27;25(17):9297. doi: 10.3390/ijms25179297. Int J Mol Sci. 2024. PMID: 39273245 Free PMC article.
-
Fine mapping with epigenetic information and 3D structure.Semin Immunopathol. 2022 Jan;44(1):115-125. doi: 10.1007/s00281-021-00906-4. Epub 2022 Jan 12. Semin Immunopathol. 2022. PMID: 35022890 Free PMC article. Review.
-
High-throughput functional dissection of noncoding SNPs with biased allelic enhancer activity for insulin resistance-relevant phenotypes.Am J Hum Genet. 2023 Aug 3;110(8):1266-1288. doi: 10.1016/j.ajhg.2023.07.002. Epub 2023 Jul 27. Am J Hum Genet. 2023. PMID: 37506691 Free PMC article.
-
Haplotype function score improves biological interpretation and cross-ancestry polygenic prediction of human complex traits.Elife. 2024 Apr 19;12:RP92574. doi: 10.7554/eLife.92574. Elife. 2024. PMID: 38639992 Free PMC article.
-
Multiple-model machine learning identifies potential functional genes in dilated cardiomyopathy.Front Cardiovasc Med. 2023 Jan 11;9:1044443. doi: 10.3389/fcvm.2022.1044443. eCollection 2022. Front Cardiovasc Med. 2023. PMID: 36712235 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
