

MicroRNA-106a Inhibits Autophagy Process and Antimicrobial Responses by Targeting ULK1, ATG7, and ATG16L1 During Mycobacterial Infection
- PMID: 33505399
- PMCID: PMC7832394
- DOI: 10.3389/fimmu.2020.610021
MicroRNA-106a Inhibits Autophagy Process and Antimicrobial Responses by Targeting ULK1, ATG7, and ATG16L1 During Mycobacterial Infection
Abstract
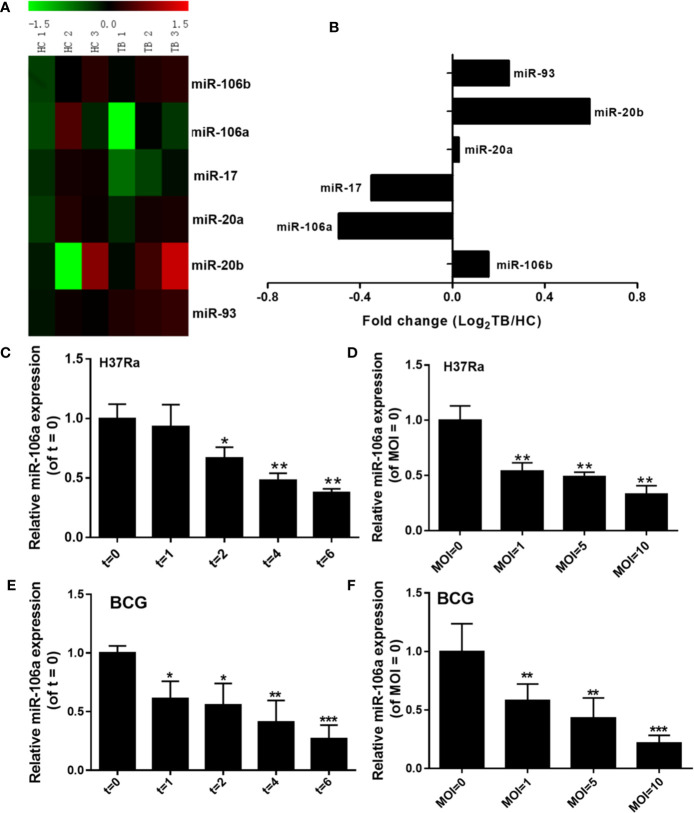
Autophagy is a key element of innate immune response against invading pathogens including Mycobacterium tuberculosis (M. tuberculosis). The emerging roles of microRNAs in regulating host antimicrobial responses against M. tuberculosis have gained widespread attention. However, the process by which miRNAs specifically influence antibacterial autophagy during mycobacterial infection is largely uncharacterized. In this study, we demonstrate a novel role of miR-106a in regulating macrophage autophagy against M. tuberculosis. H37Ra infection leads to downregulation of miR-106a in a time- and dose-dependent manner and concomitant upregulation of its three targets (ULK1, ATG7, and ATG16L1) in THP-1 macrophages. MiR-106a could inhibit autophagy activation and antimicrobial responses to M. tuberculosis by targeting ULK1, ATG7, and ATG16L1. Overexpression of miR-106a dramatically inhibited H37Ra-induced activation of autophagy in human THP-1 macrophages, whereas inhibitors of miR-106a remarkably promoted H37Ra-induced autophagy. The inhibitory effect of miR-106a on autophagy process during mycobacterial infection was also confirmed by Transmission Electron Microscope (TEM) observation. More importantly, forced expression of miR-106a increased mycobacterial survival, while transfection with miR-106a inhibitors attenuated the survival of intracellular mycobacteria. Taken together, these data demonstrated that miR-106a functioned as a negative regulator in autophagy and antimicrobial effects by targeting ULK1, ATG7, and ATG16L1 during M. tuberculosis infection, which may provide a potential target for developing diagnostic reagents or antibacterials against tuberculosis.
Keywords: ATG16L1; ATG7; Mycobacterium tuberculosis; ULK1; autophagy; miR-106a.
Copyright © 2020 Liu, Hong, Zhang, Li, He, Han, Zhang, Xu, Stonehouse, Jiang, An and Guo.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures








Similar articles
-
The Role of microRNAs and Long Non-Coding RNAs in the Regulation of the Immune Response to Mycobacterium tuberculosis Infection.Front Immunol. 2021 Jun 24;12:687962. doi: 10.3389/fimmu.2021.687962. eCollection 2021. Front Immunol. 2021. PMID: 34248974 Free PMC article. Review.
-
microRNA-20a Inhibits Autophagic Process by Targeting ATG7 and ATG16L1 and Favors Mycobacterial Survival in Macrophage Cells.Front Cell Infect Microbiol. 2016 Oct 18;6:134. doi: 10.3389/fcimb.2016.00134. eCollection 2016. Front Cell Infect Microbiol. 2016. PMID: 27803889 Free PMC article.
-
MiR-23a-5p modulates mycobacterial survival and autophagy during mycobacterium tuberculosis infection through TLR2/MyD88/NF-κB pathway by targeting TLR2.Exp Cell Res. 2017 May 15;354(2):71-77. doi: 10.1016/j.yexcr.2017.03.039. Epub 2017 Mar 19. Exp Cell Res. 2017. PMID: 28327409
-
LncRNA MIAT regulates autophagy and apoptosis of macrophage infected by Mycobacterium tuberculosis through the miR-665/ULK1 signaling axis.Mol Immunol. 2021 Nov;139:42-49. doi: 10.1016/j.molimm.2021.07.023. Epub 2021 Aug 25. Mol Immunol. 2021. PMID: 34454184
-
MicroRNA in innate immunity and autophagy during mycobacterial infection.Cell Microbiol. 2017 Jan;19(1). doi: 10.1111/cmi.12687. Epub 2016 Nov 28. Cell Microbiol. 2017. PMID: 27794209 Review.
Cited by
-
The Role of microRNAs and Long Non-Coding RNAs in the Regulation of the Immune Response to Mycobacterium tuberculosis Infection.Front Immunol. 2021 Jun 24;12:687962. doi: 10.3389/fimmu.2021.687962. eCollection 2021. Front Immunol. 2021. PMID: 34248974 Free PMC article. Review.
-
Therapeutic Relevance of Inducing Autophagy in β-Thalassemia.Cells. 2024 May 25;13(11):918. doi: 10.3390/cells13110918. Cells. 2024. PMID: 38891049 Free PMC article. Review.
-
Multi-omics study reveals differential expression and phosphorylation of autophagy-related proteins in autism spectrum disorder.Sci Rep. 2025 Mar 29;15(1):10878. doi: 10.1038/s41598-025-95860-8. Sci Rep. 2025. PMID: 40158064 Free PMC article.
-
ATG7/GAPLINC/IRF3 axis plays a critical role in regulating pathogenesis of influenza A virus.PLoS Pathog. 2024 Jan 16;20(1):e1011958. doi: 10.1371/journal.ppat.1011958. eCollection 2024 Jan. PLoS Pathog. 2024. PMID: 38227600 Free PMC article.
-
Insights on E1-like enzyme ATG7: functional regulation and relationships with aging-related diseases.Commun Biol. 2024 Mar 29;7(1):382. doi: 10.1038/s42003-024-06080-1. Commun Biol. 2024. PMID: 38553562 Free PMC article. Review.
References
-
- World Health Organization Global tuberculosis report 2020. World Health Organization (2020). Available at: https://apps.who.int/iris/handle/10665/336069. License: CC BY-NC-SA 3.0 IGO.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical