

Cancer Stemness: p53 at the Wheel
- PMID: 33505918
- PMCID: PMC7830093
- DOI: 10.3389/fonc.2020.604124
Cancer Stemness: p53 at the Wheel
Abstract
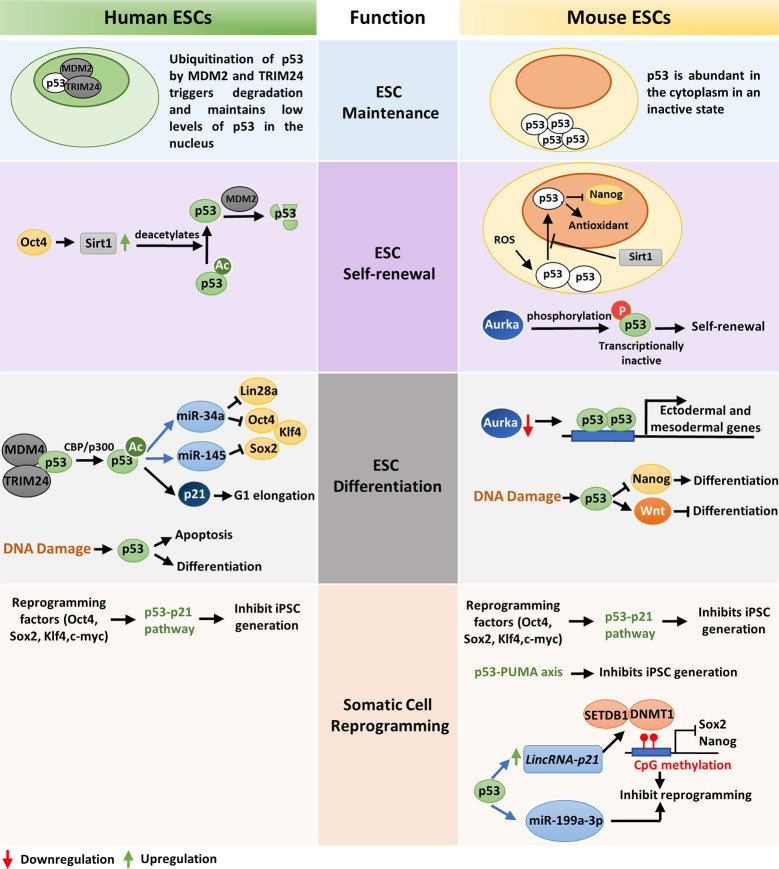
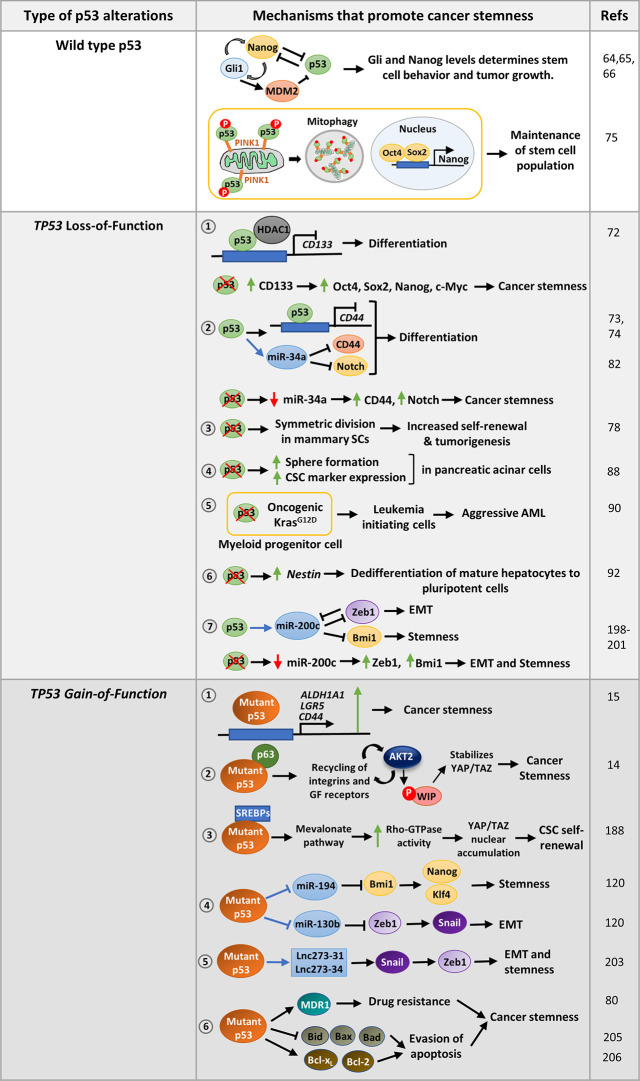
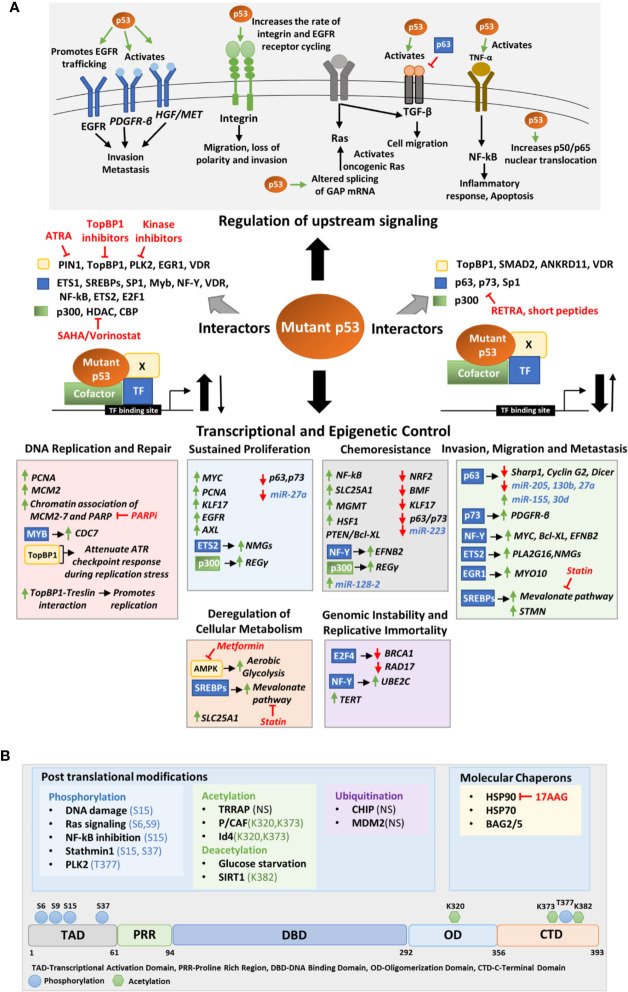
The tumor suppressor p53 maintains an equilibrium between self-renewal and differentiation to sustain a limited repertoire of stem cells for proper development and maintenance of tissue homeostasis. Inactivation of p53 disrupts this balance and promotes pluripotency and somatic cell reprogramming. A few reports in recent years have indicated that prevalent TP53 oncogenic gain-of-function (GOF) mutations further boosts the stemness properties of cancer cells. In this review, we discuss the role of wild type p53 in regulating pluripotency of normal stem cells and various mechanisms that control the balance between self-renewal and differentiation in embryonic and adult stem cells. We also highlight how inactivating and GOF mutations in p53 stimulate stemness in cancer cells. Further, we have explored the various mechanisms of mutant p53-driven cancer stemness, particularly emphasizing on the non-coding RNA mediated epigenetic regulation. We have also analyzed the association of cancer stemness with other crucial gain-of-function properties of mutant p53 such as epithelial to mesenchymal transition phenotypes and chemoresistance to understand how activation of one affects the other. Given the critical role of cancer stem-like cells in tumor maintenance, cancer progression, and therapy resistance of mutant p53 tumors, targeting them might improve therapeutic efficacy in human cancers with TP53 mutations.
Keywords: GOF mutant p53; cancer stemness; chemoresistance; differentiation; epithelial to mesenchymal transition; miRNAs; therapeutic targeting.
Copyright © 2021 Ghatak, Das Ghosh and Roychoudhury.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous