

Early Diagnosis and Intervention in Cystic Fibrosis: Imagining the Unimaginable
- PMID: 33505947
- PMCID: PMC7830672
- DOI: 10.3389/fped.2020.608821
Early Diagnosis and Intervention in Cystic Fibrosis: Imagining the Unimaginable
Abstract
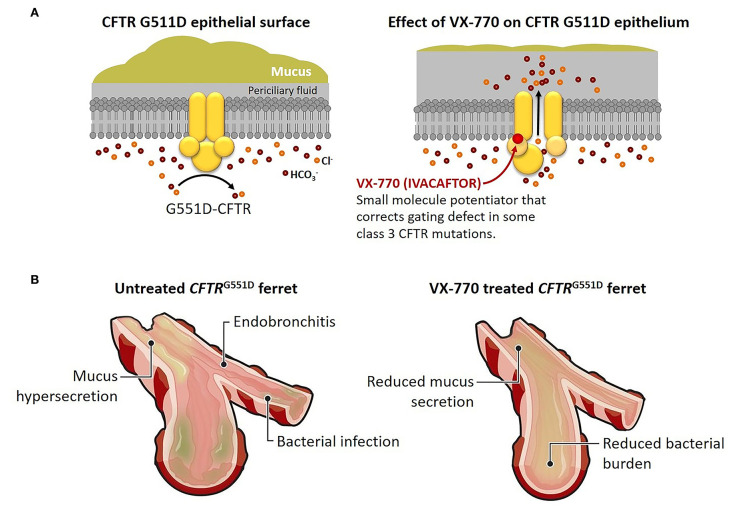
Cystic fibrosis is the most common life-shortening genetic disease affecting Caucasians, clinically manifested by fat malabsorption, poor growth and nutrition, and recurrent sinopulmonary infections. Newborn screening programs for cystic fibrosis are now implemented throughout the United States and in many nations worldwide. Early diagnosis and interventions have led to improved clinical outcomes for people with cystic fibrosis. Newer cystic fibrosis transmembrane conductance regulator potentiators and correctors with mutation-specific effects have increasingly been used in children, and these agents are revolutionizing care. Indeed, it is possible that highly effective modulator therapy used early in life could profoundly affect the trajectory of cystic fibrosis lung disease, and primary prevention may be achievable.
Keywords: corrector; cystic fibrosis; cystic fibrosis transmembrane conductance regulator; immunoreactive trypsin(ogen); newborn screening; potentiator; sweat chloride test.
Copyright © 2021 Coverstone and Ferkol.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Guthrie R, Susi A. A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants. Pediatrics. (1963) 32:338–43. - PubMed
-
- Wilson JMG, Jungner G. Principles and practice of screening for disease. Who Chron. (1968) 22:473.
-
- Michelson P, Faro A, Ferkol T. Pulmonary disease in cystic fibrosis. In: Wilmott R, Bush A, Ratjen F, et al. editors. Kendig's Disorders of the Respiratory Tract in Children. 9th Edn Philadelphia, PA: Elsevier Health Sciences; (2018). p. 777–87.
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources