

Therapeutic targeting of argininosuccinate synthase 1 (ASS1)-deficient pulmonary fibrosis
- PMID: 33508432
- PMCID: PMC8058484
- DOI: 10.1016/j.ymthe.2021.01.028
Therapeutic targeting of argininosuccinate synthase 1 (ASS1)-deficient pulmonary fibrosis
Abstract
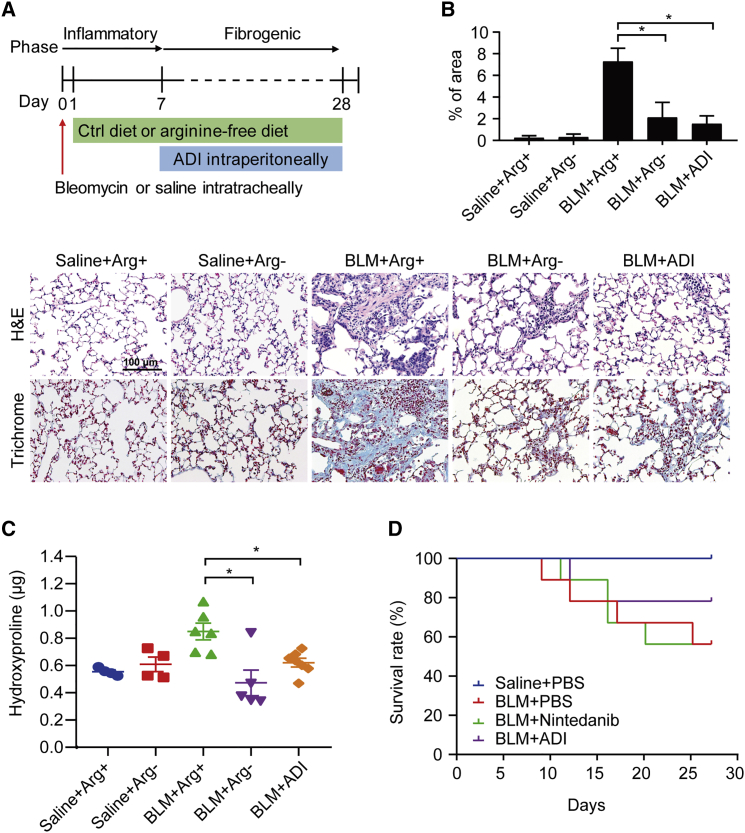
Argininosuccinate synthase 1 (ASS1) serves as a critical enzyme in arginine biosynthesis; however, its role in interstitial lung diseases, particularly idiopathic pulmonary fibrosis (IPF), remains largely unknown. This study aims at characterization and targeting of ASS1 deficiency in pulmonary fibrosis. We find that ASS1 was significantly decreased and inversely correlated with fibrotic status. Transcriptional downregulation of ASS1 was noted in fibroblastic foci of primary lung fibroblasts isolated from IPF patients. Genetic manipulations of ASS1 studies confirm that ASS1 expression inhibited fibroblast cell proliferation, migration, and invasion. We further show that the hepatocyte growth factor receptor (Met) receptor was activated and acted upstream of the Src-STAT3 axis signaling in ASS1-knockdown fibroblasts. Interestingly, both arginine-free conditions and arginine deiminase treatment were demonstrated to kill fibrotic fibroblasts, attenuated bleomycin-induced pulmonary fibrosis in mice, as well as synergistically increased nintedanib efficacy. Our data suggest ASS1 deficiency as a druggable target and also provide a unique therapeutic strategy against pulmonary fibrosis.
Keywords: ASS1; MET signaling; arginine; fibroblasts; pulmonary fibrosis.
Copyright © 2021 The American Society of Gene and Cell Therapy. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare no competing interests.
Figures






Comment in
-
Arginine is a key player in fibroblasts during the course of IPF development.Mol Ther. 2021 Apr 7;29(4):1361-1363. doi: 10.1016/j.ymthe.2021.02.023. Epub 2021 Feb 26. Mol Ther. 2021. PMID: 33639113 Free PMC article. No abstract available.
Similar articles
-
Inositol possesses antifibrotic activity and mitigates pulmonary fibrosis.Respir Res. 2023 May 16;24(1):132. doi: 10.1186/s12931-023-02421-6. Respir Res. 2023. PMID: 37194070 Free PMC article.
-
Argininosuccinate synthetase 1 suppression and arginine restriction inhibit cell migration in gastric cancer cell lines.Sci Rep. 2015 Apr 30;5:9783. doi: 10.1038/srep09783. Sci Rep. 2015. PMID: 25928182 Free PMC article.
-
[Shenqi Chongcao Formula ameliorates inflammatory response in rats with pulmonary fibrosis by activating the ASS1/src/STAT3 signaling pathway].Nan Fang Yi Ke Da Xue Xue Bao. 2024 Apr 20;44(4):644-651. doi: 10.12122/j.issn.1673-4254.2024.04.04. Nan Fang Yi Ke Da Xue Xue Bao. 2024. PMID: 38708496 Free PMC article. Chinese.
-
Citrullinemia type I is associated with a novel splicing variant, c.773 + 4A > C, in ASS1: a case report and literature review.BMC Med Genet. 2019 Jun 17;20(1):110. doi: 10.1186/s12881-019-0836-5. BMC Med Genet. 2019. PMID: 31208364 Free PMC article. Review.
-
Mutations and polymorphisms in the human argininosuccinate synthetase (ASS1) gene.Hum Mutat. 2009 Mar;30(3):300-7. doi: 10.1002/humu.20847. Hum Mutat. 2009. PMID: 19006241 Review.
Cited by
-
Metabolic dysregulation in pulmonary fibrosis: insights into amino acid contributions and therapeutic potential.Cell Death Discov. 2025 Aug 27;11(1):411. doi: 10.1038/s41420-025-02715-2. Cell Death Discov. 2025. PMID: 40858556 Free PMC article. Review.
-
Spatial and Single-Cell Transcriptomics Reveals the Regional Division of the Spatial Structure of MASH Fibrosis.Liver Int. 2025 Apr;45(4):e16125. doi: 10.1111/liv.16125. Epub 2024 Oct 14. Liver Int. 2025. PMID: 39400982 Free PMC article.
-
Arginine promotes the activation of human lung fibroblasts independent of its metabolism.Biochem J. 2025 Jun 17;482(12):823-38. doi: 10.1042/BCJ20253033. Biochem J. 2025. PMID: 40454944 Free PMC article.
-
A novel gene signature based on the hub genes of COVID-19 predicts the prognosis of idiopathic pulmonary fibrosis.Front Pharmacol. 2022 Sep 6;13:981604. doi: 10.3389/fphar.2022.981604. eCollection 2022. Front Pharmacol. 2022. PMID: 36147332 Free PMC article.
-
Identifying a survival-associated cell type based on multi-level transcriptome analysis in idiopathic pulmonary fibrosis.Respir Res. 2024 Mar 15;25(1):126. doi: 10.1186/s12931-024-02738-w. Respir Res. 2024. PMID: 38491375 Free PMC article.
References
-
- Ley B., Collard H.R., King T.E., Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2011;183:431–440. - PubMed
-
- Martinez F.J., Collard H.R., Pardo A., Raghu G., Richeldi L., Selman M., Swigris J.J., Taniguchi H., Wells A.U. Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Primers. 2017;3:17074. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
