

Noncanonical open reading frames encode functional proteins essential for cancer cell survival
- PMID: 33510483
- PMCID: PMC8195866
- DOI: 10.1038/s41587-020-00806-2
Noncanonical open reading frames encode functional proteins essential for cancer cell survival
Abstract
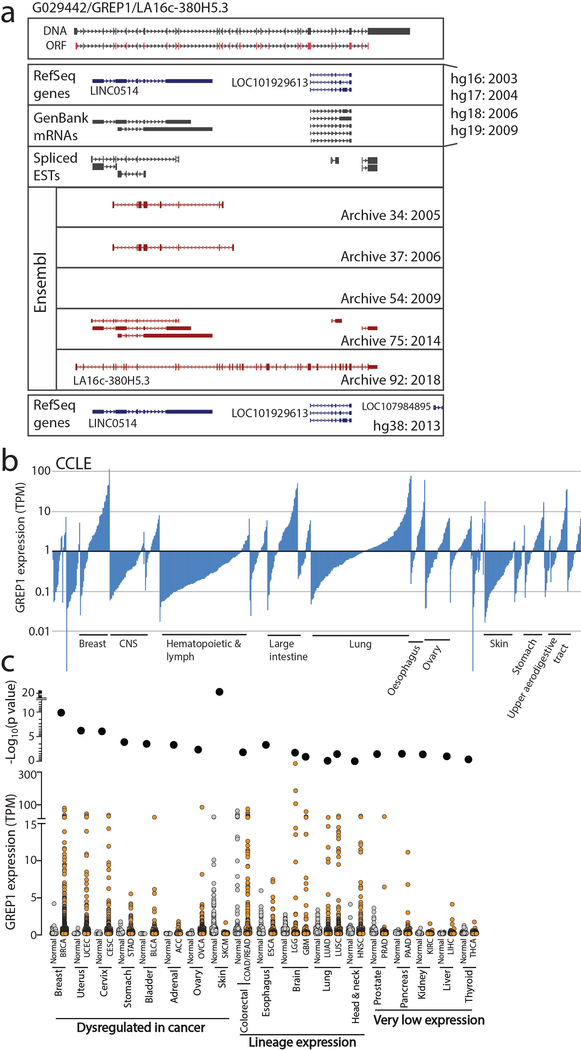
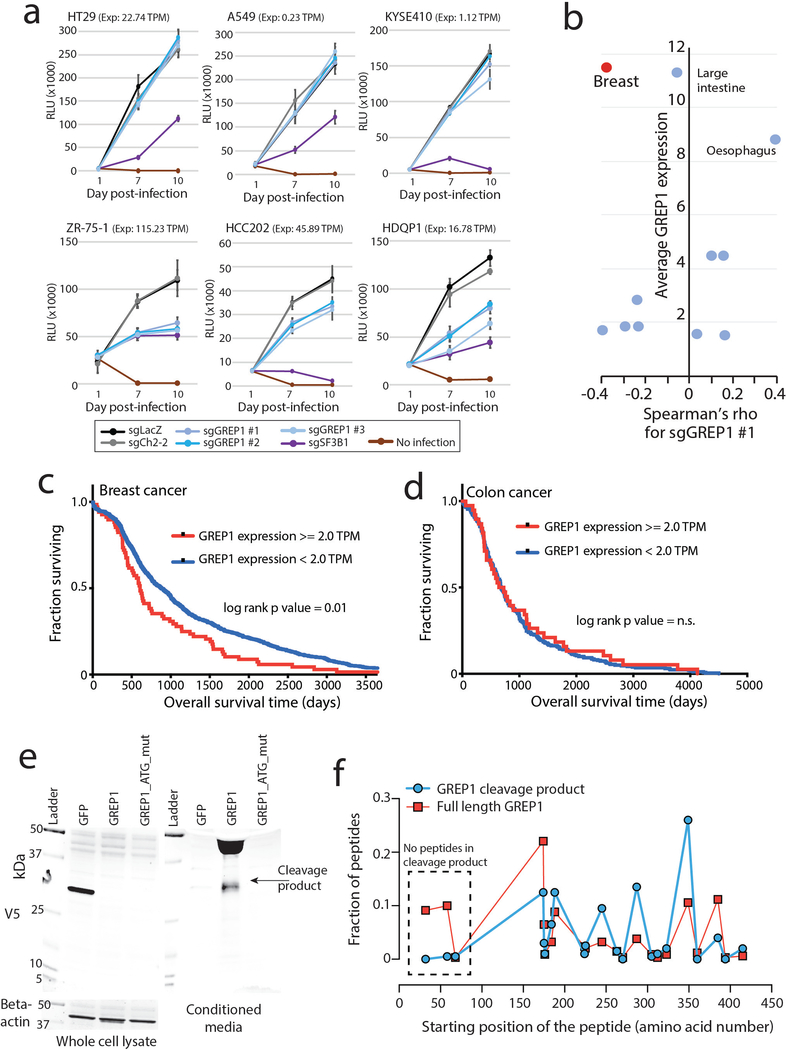
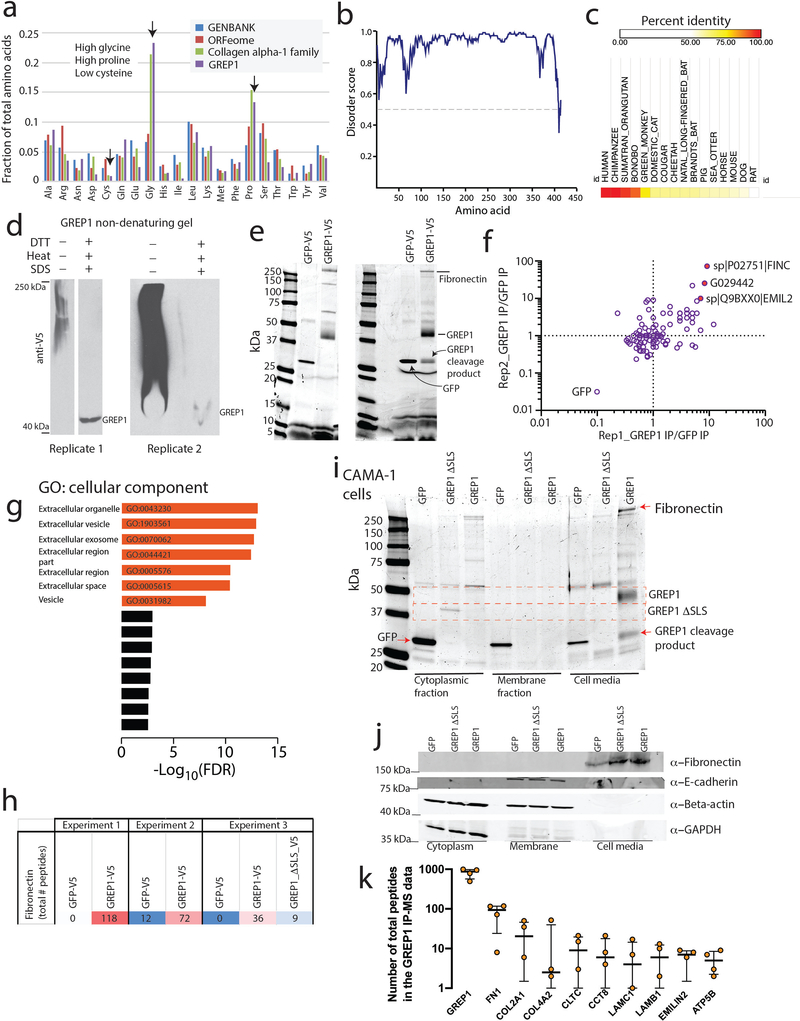
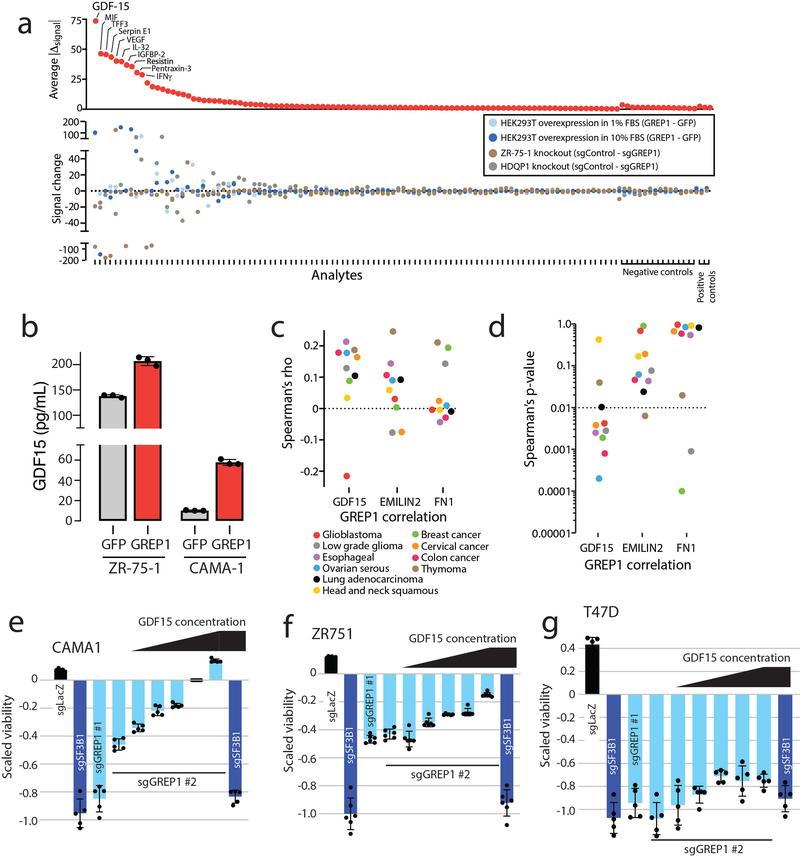
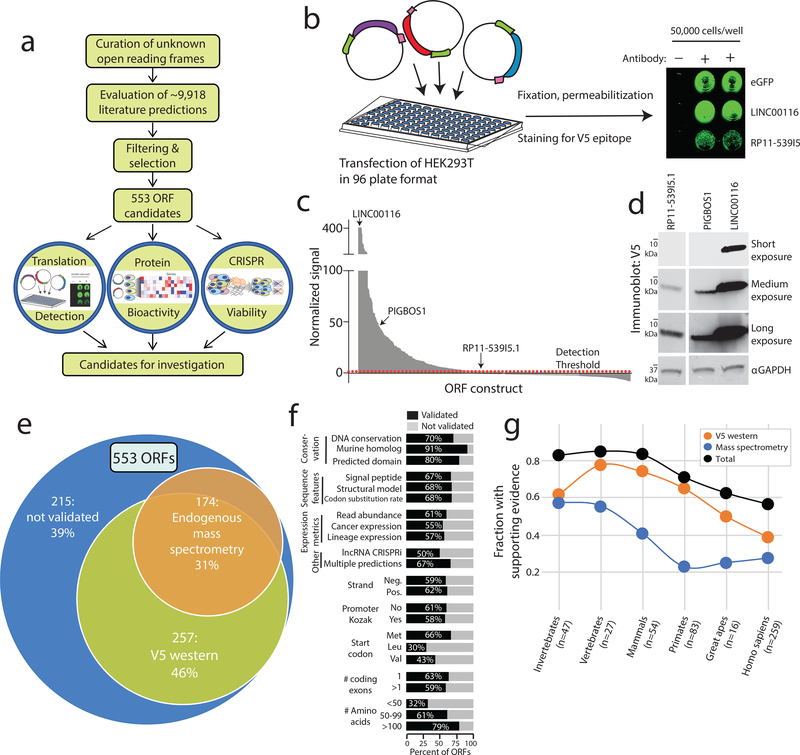
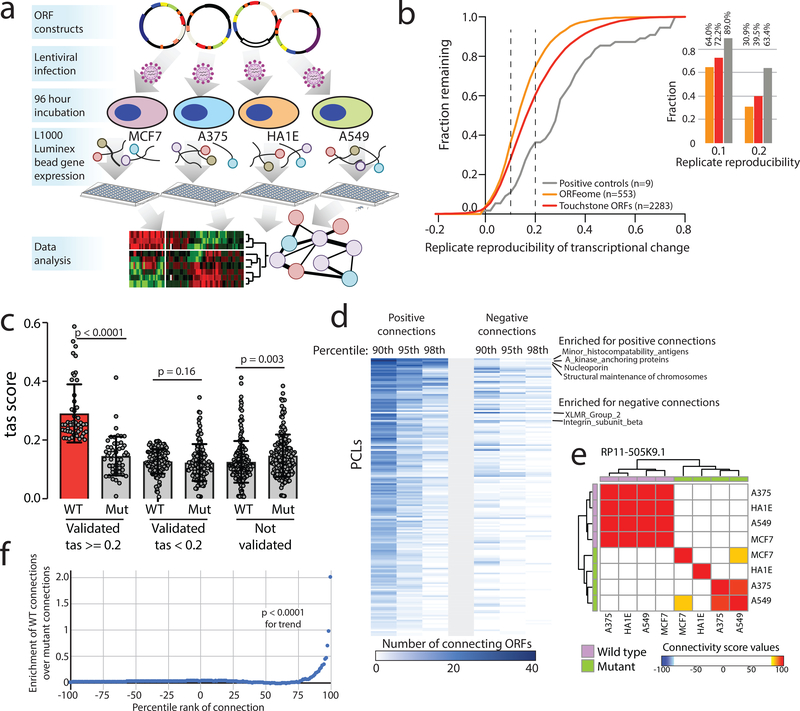
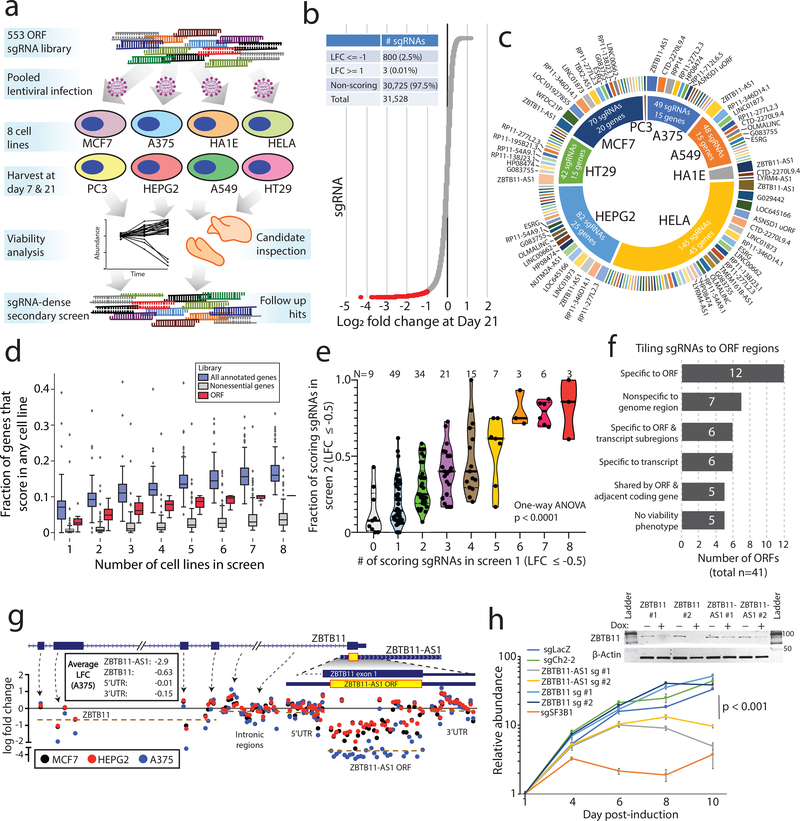
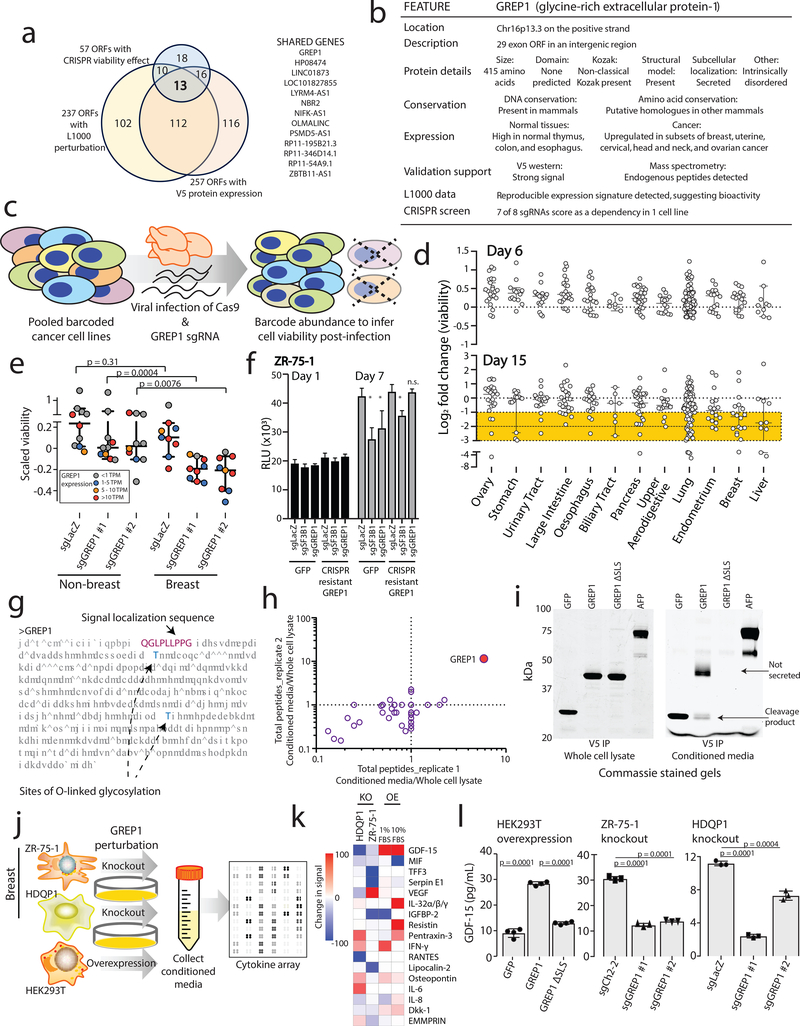
Although genomic analyses predict many noncanonical open reading frames (ORFs) in the human genome, it is unclear whether they encode biologically active proteins. Here we experimentally interrogated 553 candidates selected from noncanonical ORF datasets. Of these, 57 induced viability defects when knocked out in human cancer cell lines. Following ectopic expression, 257 showed evidence of protein expression and 401 induced gene expression changes. Clustered regularly interspaced short palindromic repeat (CRISPR) tiling and start codon mutagenesis indicated that their biological effects required translation as opposed to RNA-mediated effects. We found that one of these ORFs, G029442-renamed glycine-rich extracellular protein-1 (GREP1)-encodes a secreted protein highly expressed in breast cancer, and its knockout in 263 cancer cell lines showed preferential essentiality in breast cancer-derived lines. The secretome of GREP1-expressing cells has an increased abundance of the oncogenic cytokine GDF15, and GDF15 supplementation mitigated the growth-inhibitory effect of GREP1 knockout. Our experiments suggest that noncanonical ORFs can express biologically active proteins that are potential therapeutic targets.
Conflict of interest statement
Competing Interests Statement
The authors declare no relevant competing interests.
Figures
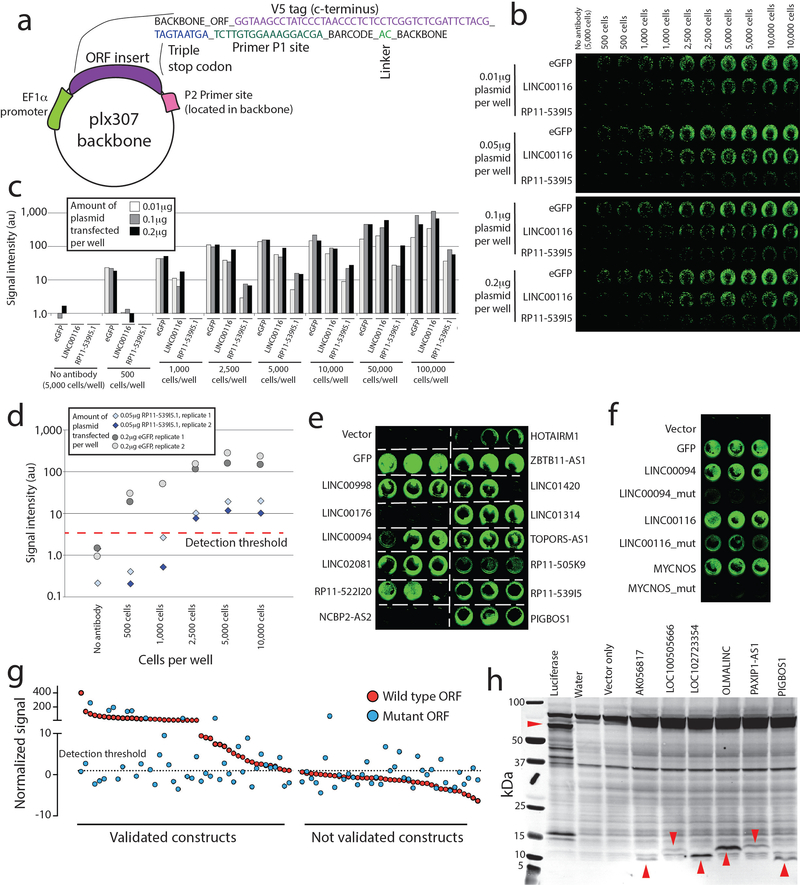
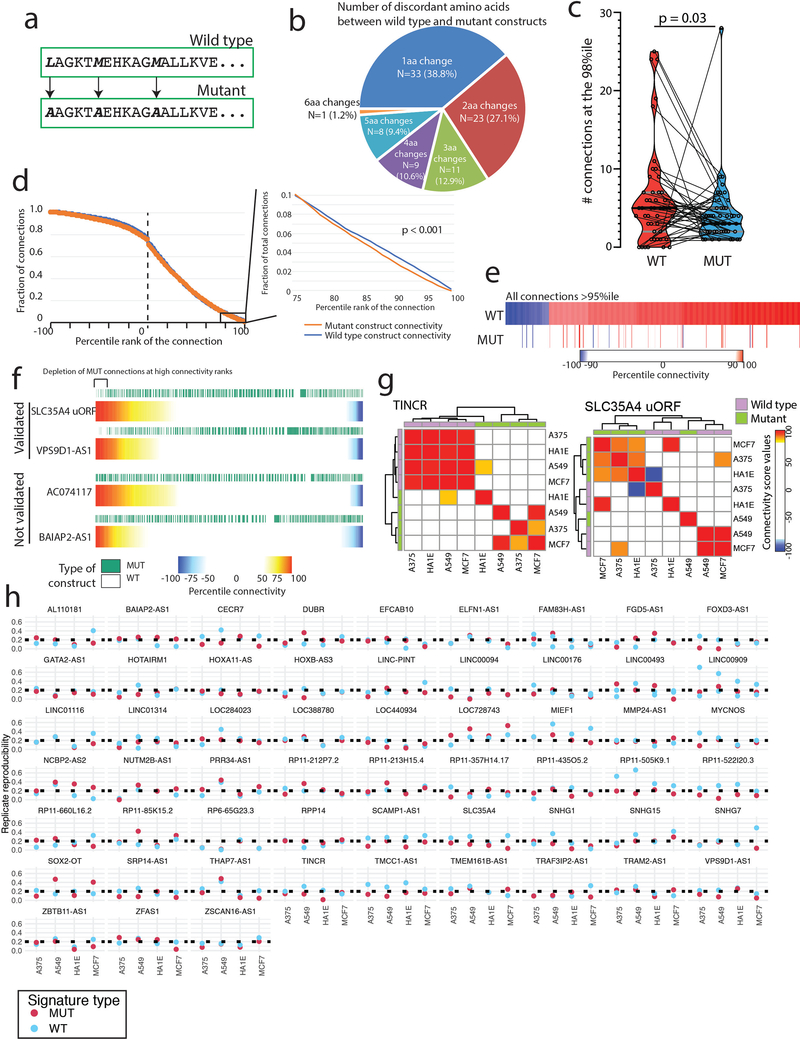
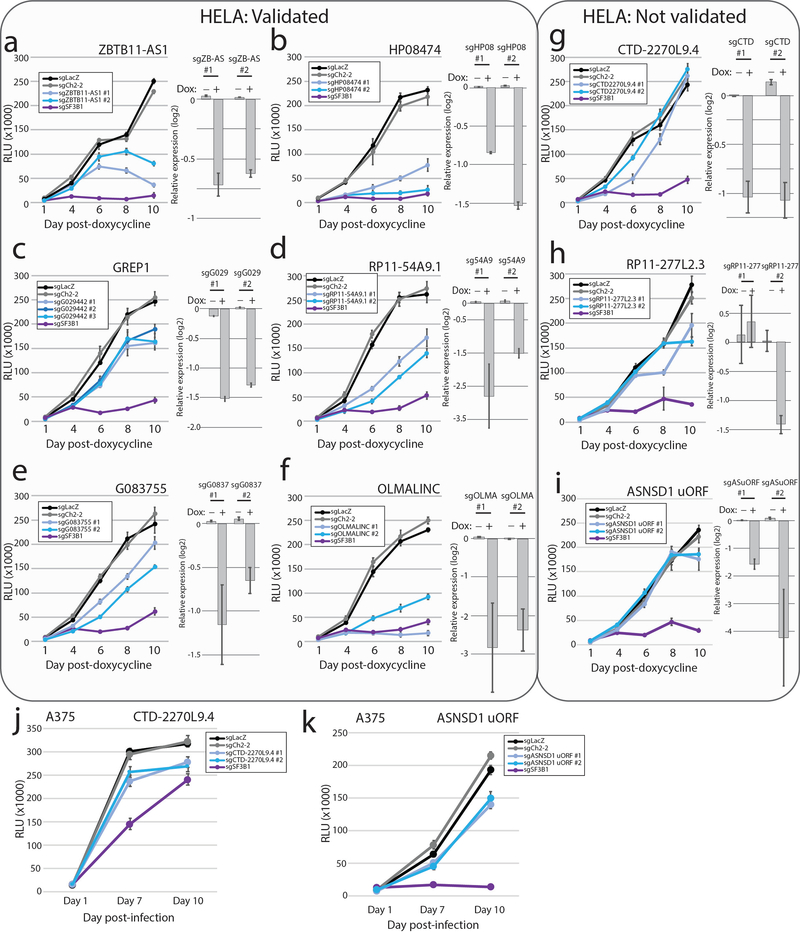

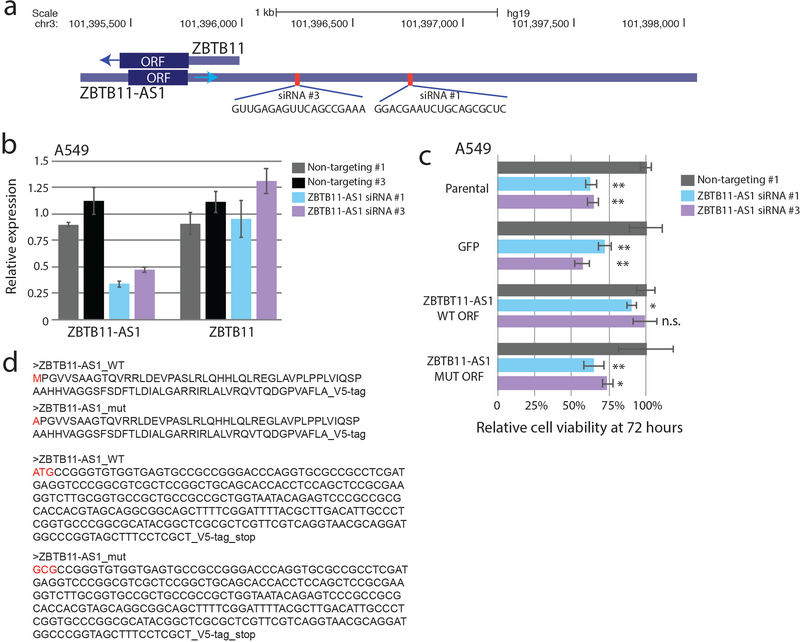
References
-
- Ewing B & Green P Analysis of expressed sequence tags indicates 35,000 human genes. Nat. Genet 25, 232–234 (2000). - PubMed
-
- Fields C, Adams MD, White O & Venter JC How many genes in the human genome? Nat. Genet 7, 345–346 (1994). - PubMed
-
- Liang F et al. Gene index analysis of the human genome estimates approximately 120,000 genes. Nat. Genet 25, 239–240 (2000). - PubMed
Methods-Only References
-
- Petersen TN, Brunak S, von Heijne G & Nielsen H SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786 (2011). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
