

Dynamics of the HD regulatory subdomain of PARP-1; substrate access and allostery in PARP activation and inhibition
- PMID: 33511412
- PMCID: PMC7913765
- DOI: 10.1093/nar/gkab020
Dynamics of the HD regulatory subdomain of PARP-1; substrate access and allostery in PARP activation and inhibition
Abstract
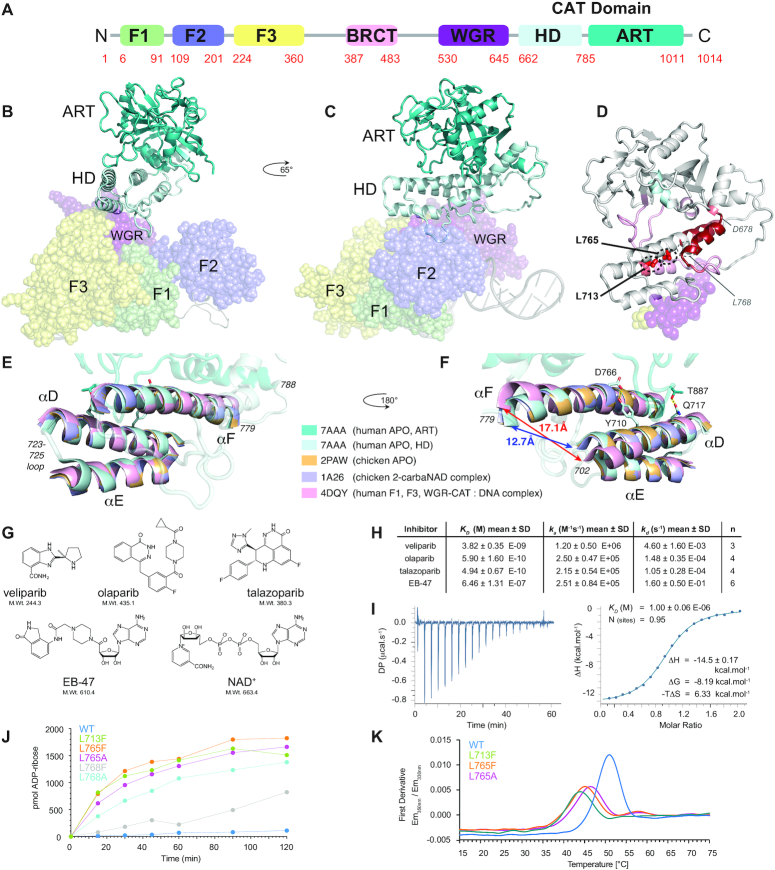
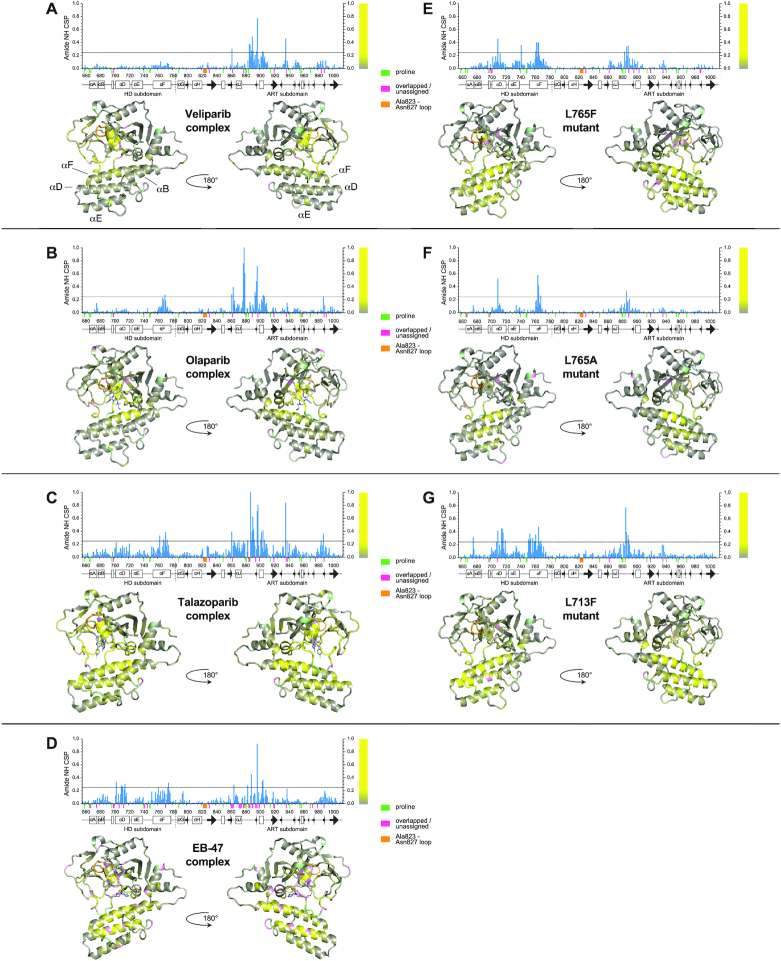
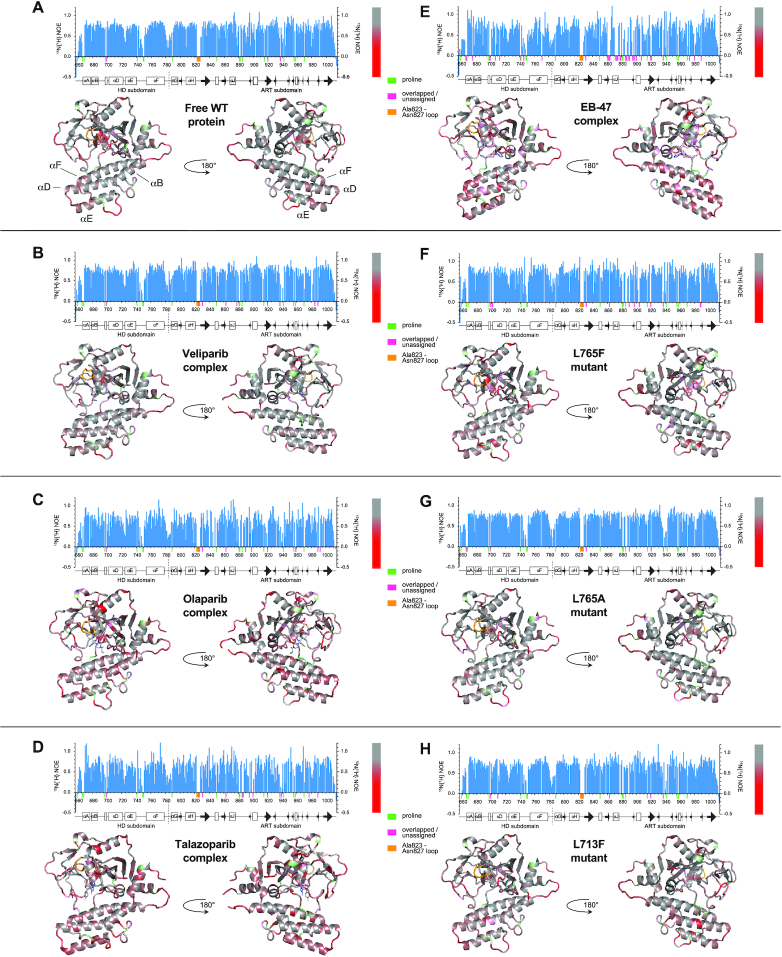
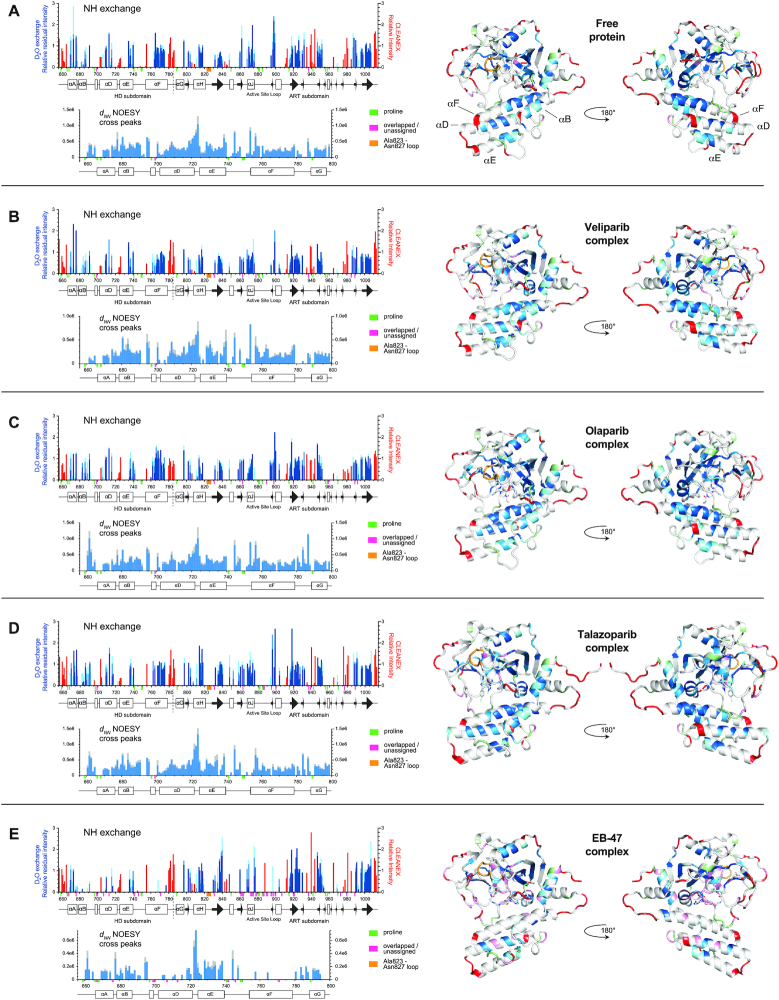
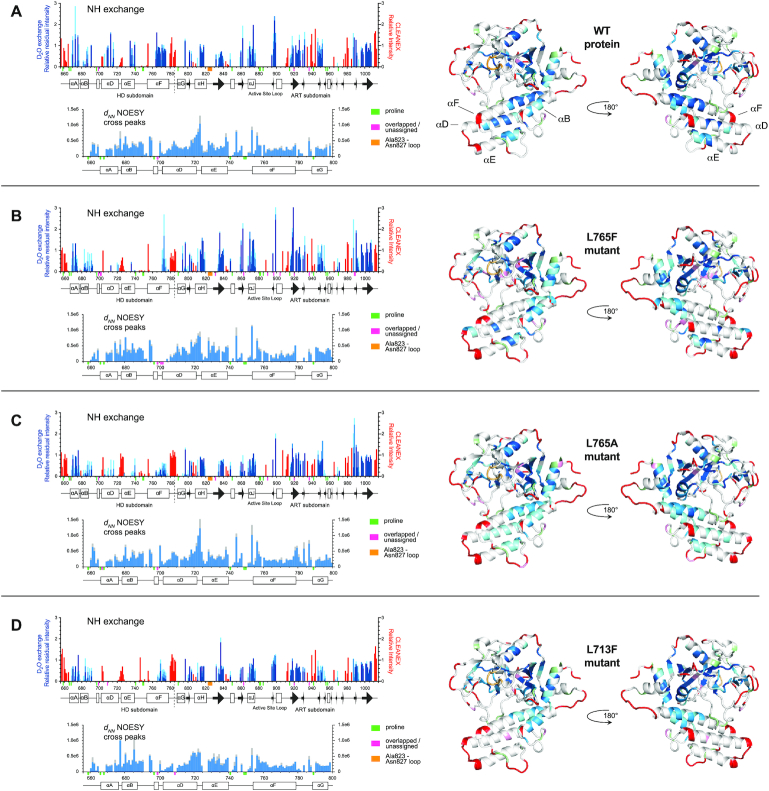
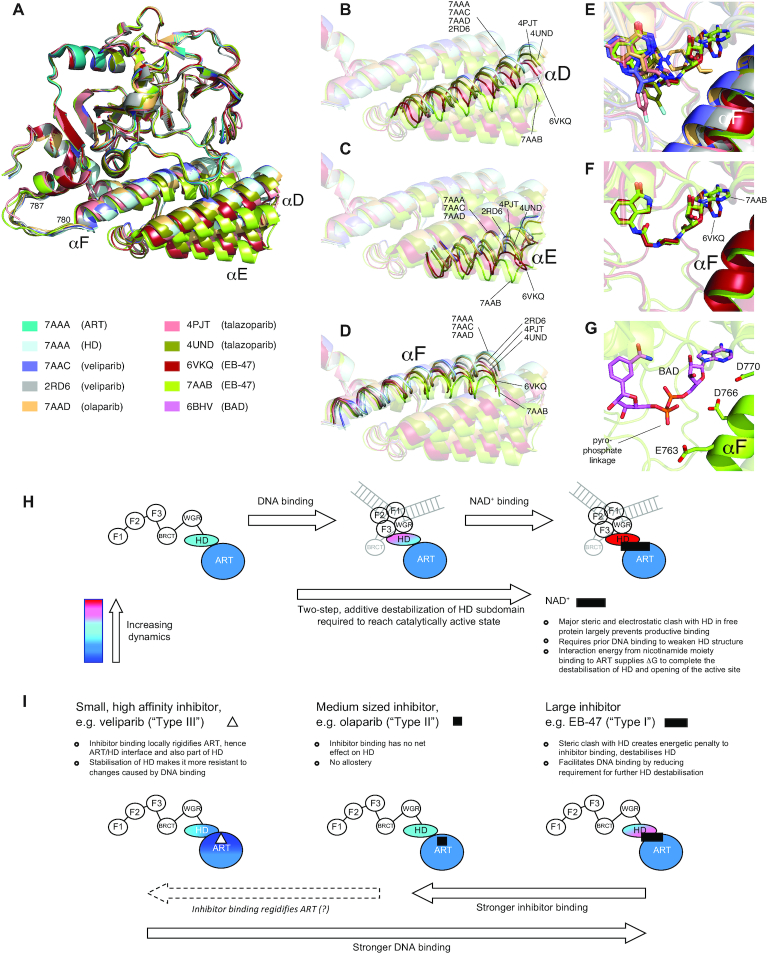
PARP-1 is a key early responder to DNA damage in eukaryotic cells. An allosteric mechanism links initial sensing of DNA single-strand breaks by PARP-1's F1 and F2 domains via a process of further domain assembly to activation of the catalytic domain (CAT); synthesis and attachment of poly(ADP-ribose) (PAR) chains to protein sidechains then signals for assembly of DNA repair components. A key component in transmission of the allosteric signal is the HD subdomain of CAT, which alone bridges between the assembled DNA-binding domains and the active site in the ART subdomain of CAT. Here we present a study of isolated CAT domain from human PARP-1, using NMR-based dynamics experiments to analyse WT apo-protein as well as a set of inhibitor complexes (with veliparib, olaparib, talazoparib and EB-47) and point mutants (L713F, L765A and L765F), together with new crystal structures of the free CAT domain and inhibitor complexes. Variations in both dynamics and structures amongst these species point to a model for full-length PARP-1 activation where first DNA binding and then substrate interaction successively destabilise the folded structure of the HD subdomain to the point where its steric blockade of the active site is released and PAR synthesis can proceed.
© The Author(s) 2021. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
Similar articles
-
Captured snapshots of PARP1 in the active state reveal the mechanics of PARP1 allostery.Mol Cell. 2022 Aug 18;82(16):2939-2951.e5. doi: 10.1016/j.molcel.2022.06.011. Epub 2022 Jul 5. Mol Cell. 2022. PMID: 35793673 Free PMC article.
-
PARP Power: A Structural Perspective on PARP1, PARP2, and PARP3 in DNA Damage Repair and Nucleosome Remodelling.Int J Mol Sci. 2021 May 12;22(10):5112. doi: 10.3390/ijms22105112. Int J Mol Sci. 2021. PMID: 34066057 Free PMC article. Review.
-
Structural basis for allosteric PARP-1 retention on DNA breaks.Science. 2020 Apr 3;368(6486):eaax6367. doi: 10.1126/science.aax6367. Science. 2020. PMID: 32241924 Free PMC article.
-
Fluorescent sensors of PARP-1 structural dynamics and allosteric regulation in response to DNA damage.Nucleic Acids Res. 2016 Nov 16;44(20):9771-9783. doi: 10.1093/nar/gkw710. Epub 2016 Aug 16. Nucleic Acids Res. 2016. PMID: 27530425 Free PMC article.
-
Poly(ADP-ribose) Polymerase (PARP) and PARP Inhibitors: Mechanisms of Action and Role in Cardiovascular Disorders.Cardiovasc Toxicol. 2018 Dec;18(6):493-506. doi: 10.1007/s12012-018-9462-2. Cardiovasc Toxicol. 2018. PMID: 29968072 Review.
Cited by
-
Single-molecule force spectroscopy reveals binding and bridging dynamics of PARP1 and PARP2 at DNA double-strand breaks.Proc Natl Acad Sci U S A. 2023 May 30;120(22):e2214209120. doi: 10.1073/pnas.2214209120. Epub 2023 May 22. Proc Natl Acad Sci U S A. 2023. PMID: 37216533 Free PMC article.
-
A Tandem-Affinity Purification Method for Identification of Primary Intracellular Drug-Binding Proteins.ACS Chem Biol. 2024 Feb 16;19(2):233-242. doi: 10.1021/acschembio.3c00570. Epub 2024 Jan 25. ACS Chem Biol. 2024. PMID: 38271588 Free PMC article.
-
Updated protein domain annotation of the PARP protein family sheds new light on biological function.Nucleic Acids Res. 2023 Aug 25;51(15):8217-8236. doi: 10.1093/nar/gkad514. Nucleic Acids Res. 2023. PMID: 37326024 Free PMC article.
-
Dual function of HPF1 in the modulation of PARP1 and PARP2 activities.Commun Biol. 2021 Nov 3;4(1):1259. doi: 10.1038/s42003-021-02780-0. Commun Biol. 2021. PMID: 34732825 Free PMC article.
-
A two-step mechanism governing PARP1-DNA retention by PARP inhibitors.Sci Adv. 2022 Sep 9;8(36):eabq0414. doi: 10.1126/sciadv.abq0414. Epub 2022 Sep 7. Sci Adv. 2022. PMID: 36070389 Free PMC article.
References
-
- Caldecott K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008; 9:619–631. - PubMed
-
- Bryant H.E., Schultz N., Thomas H.D., Parker K.M., Flower D., Lopez E., Kyle S., Meuth M., Curtin N.J., Helleday T.. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005; 434:913–917. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
