

DNA damage-how and why we age?
- PMID: 33512317
- PMCID: PMC7846274
- DOI: 10.7554/eLife.62852
DNA damage-how and why we age?
Abstract
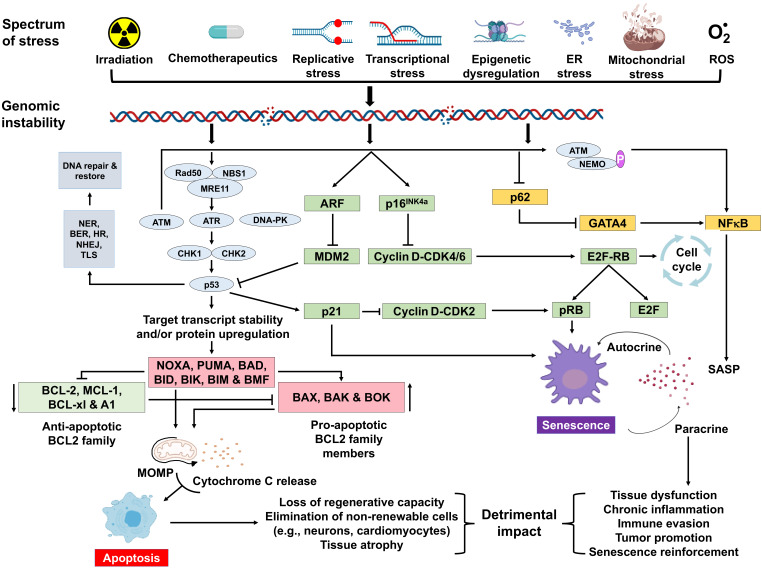
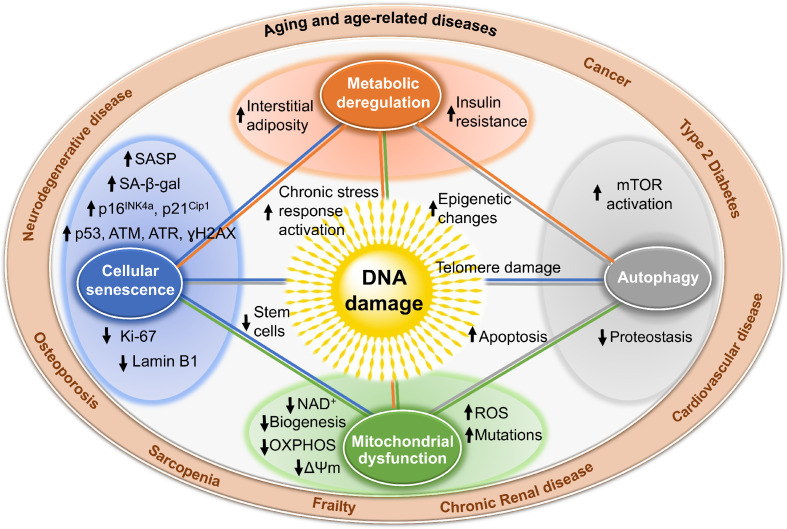
Aging is a complex process that results in loss of the ability to reattain homeostasis following stress, leading, thereby, to increased risk of morbidity and mortality. Many factors contribute to aging, such as the time-dependent accumulation of macromolecular damage, including DNA damage. The integrity of the nuclear genome is essential for cellular, tissue, and organismal health. DNA damage is a constant threat because nucleic acids are chemically unstable under physiological conditions and vulnerable to attack by endogenous and environmental factors. To combat this, all organisms possess highly conserved mechanisms to detect and repair DNA damage. Persistent DNA damage (genotoxic stress) triggers signaling cascades that drive cells into apoptosis or senescence to avoid replicating a damaged genome. The drawback is that these cancer avoidance mechanisms promote aging. Here, we review evidence that DNA damage plays a causal role in aging. We also provide evidence that genotoxic stress is linked to other cellular processes implicated as drivers of aging, including mitochondrial and metabolic dysfunction, altered proteostasis and inflammation. These links between damage to the genetic code and other pillars of aging support the notion that DNA damage could be the root of aging.
Keywords: Aging; DNA damage; DNA repair; genetics; genome instability; genomics; progeria.
© 2021, Yousefzadeh et al.
Conflict of interest statement
MY, CH, RV, CS, PR, LN No competing interests declared
Figures
References
-
- Alimirah F, Pulido T, Valdovinos A, Alptekin S, Chang E, Jones E, Diaz DA, Flores J, Velarde MC, Demaria M, Davalos AR, Wiley CD, Limbad C, Desprez PY, Campisi J. Cellular senescence promotes skin carcinogenesis through p38MAPK and p44/42MAPK signaling. Cancer Research. 2020;80:3606–3619. doi: 10.1158/0008-5472.CAN-20-0108. - DOI - PMC - PubMed
-
- Armenian SH, Armstrong GT, Aune G, Chow EJ, Ehrhardt MJ, Ky B, Moslehi J, Mulrooney DA, Nathan PC, Ryan TD, van der Pal HJ, van Dalen EC, Kremer LCM. Cardiovascular disease in survivors of childhood Cancer: insights into epidemiology, pathophysiology, and prevention. Journal of Clinical Oncology. 2018;36:2135–2144. doi: 10.1200/JCO.2017.76.3920. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
