

Diagnostic blood RNA profiles for human acute spinal cord injury
- PMID: 33512429
- PMCID: PMC7852457
- DOI: 10.1084/jem.20201795
Diagnostic blood RNA profiles for human acute spinal cord injury
Abstract
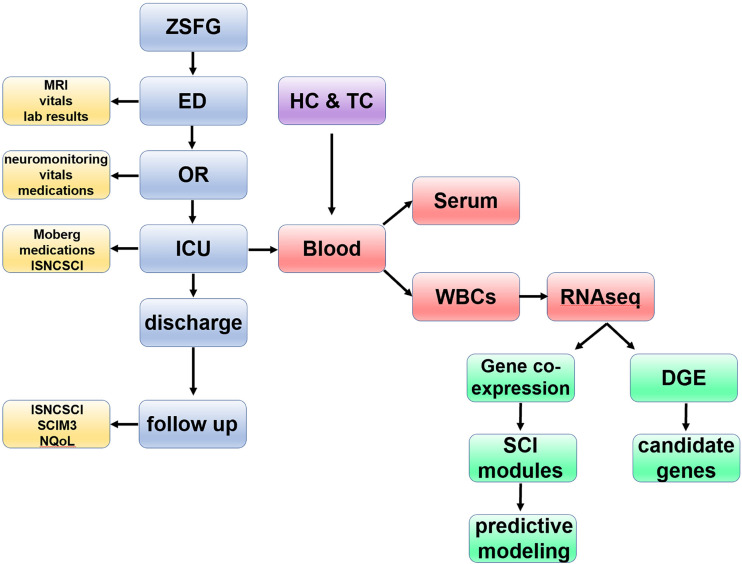
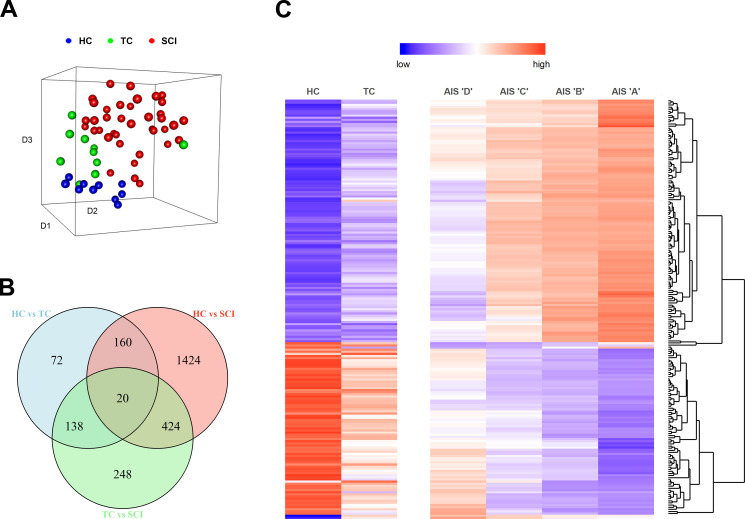
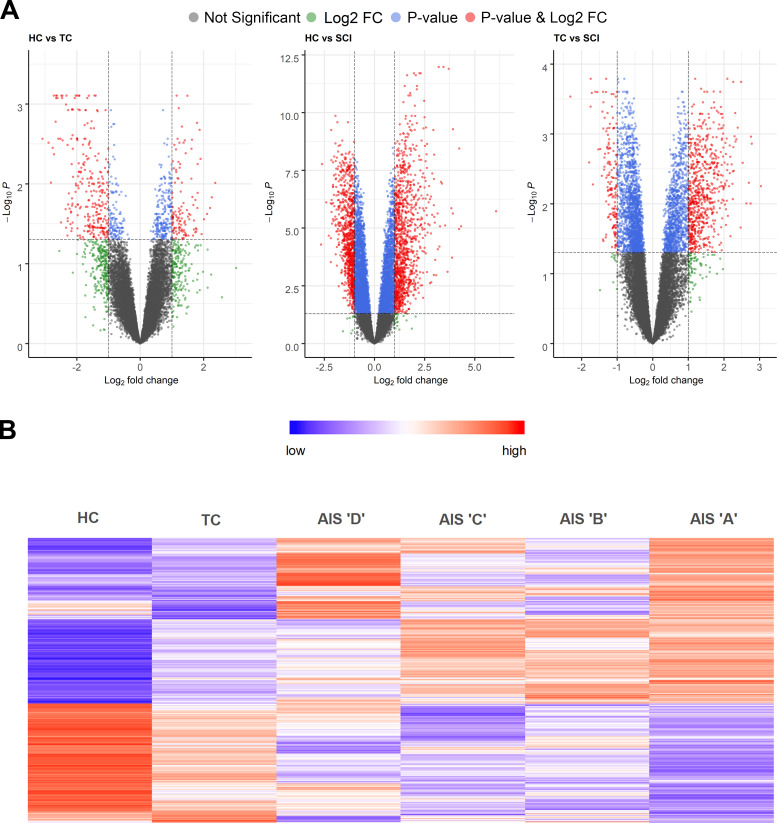
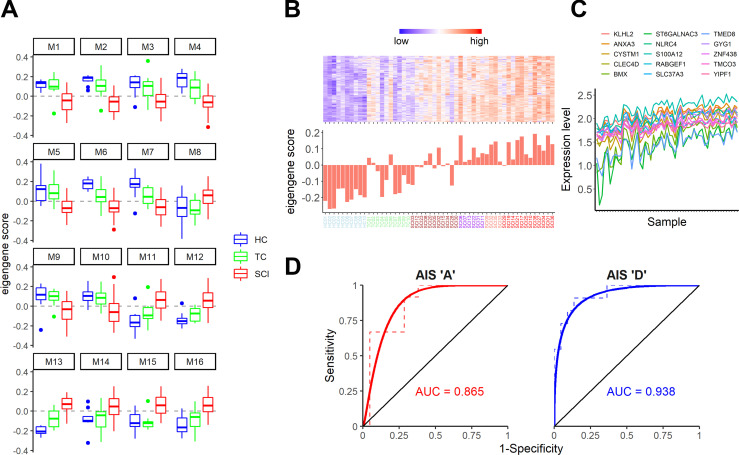
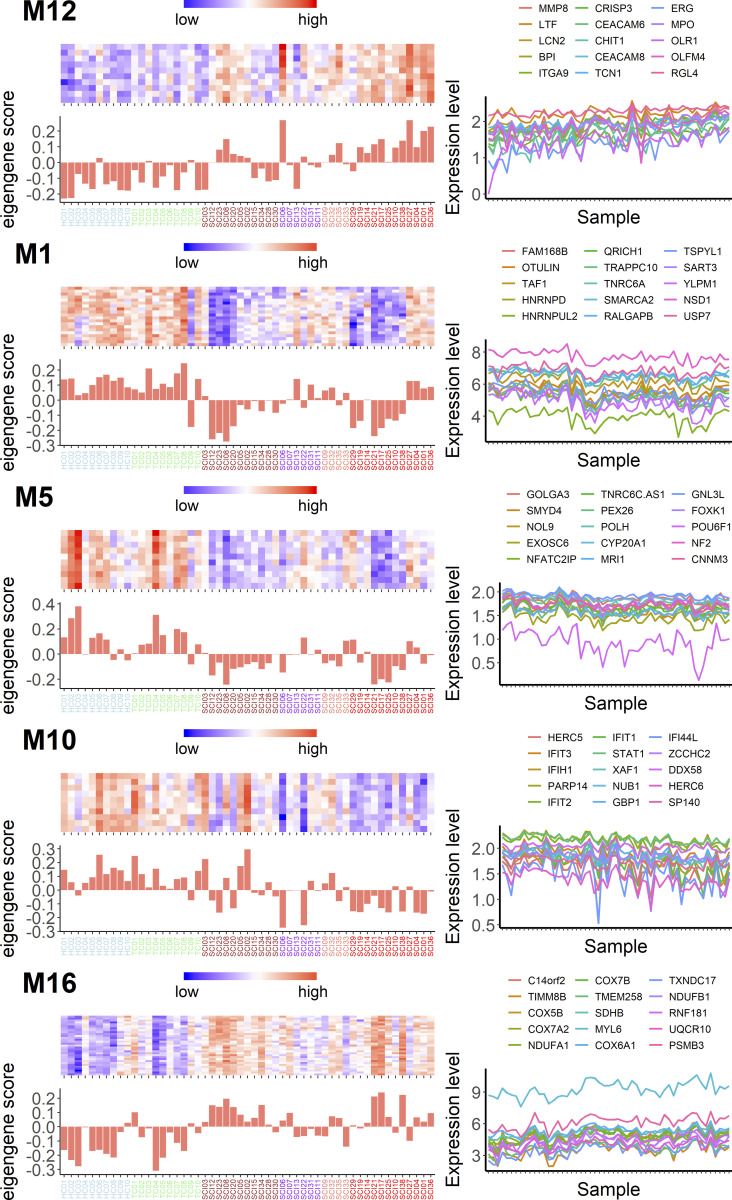
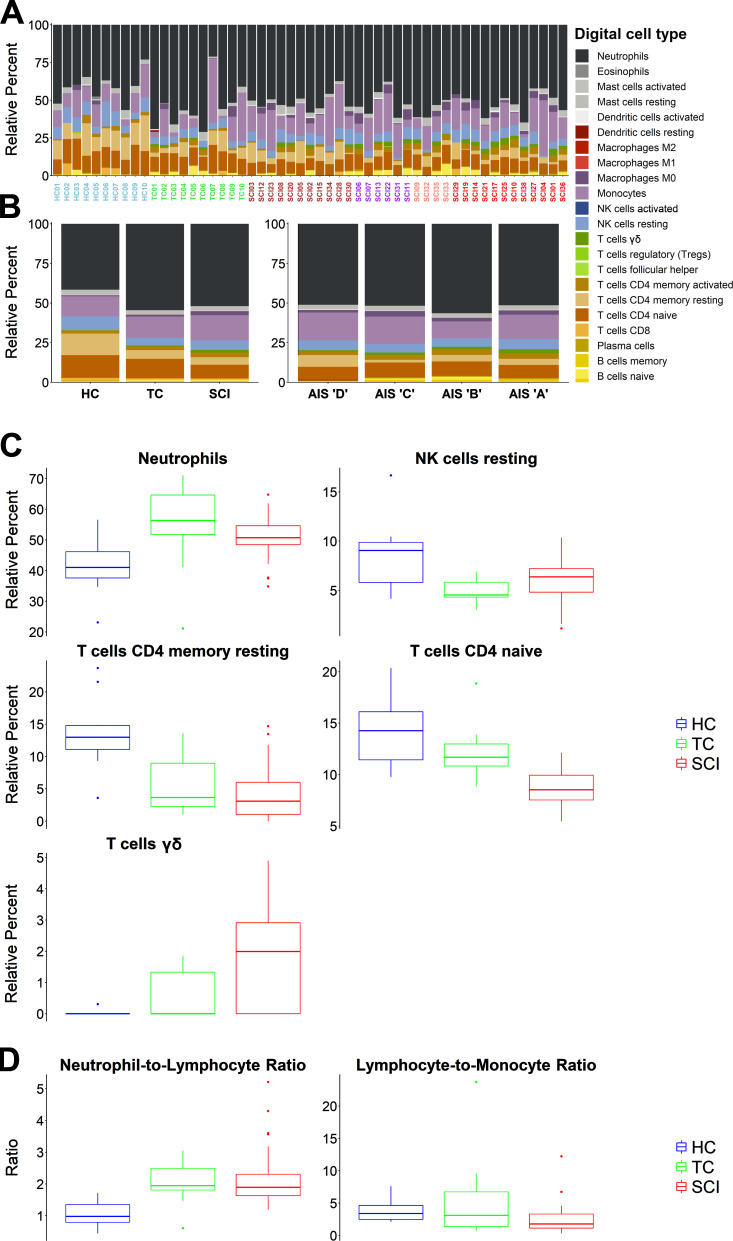
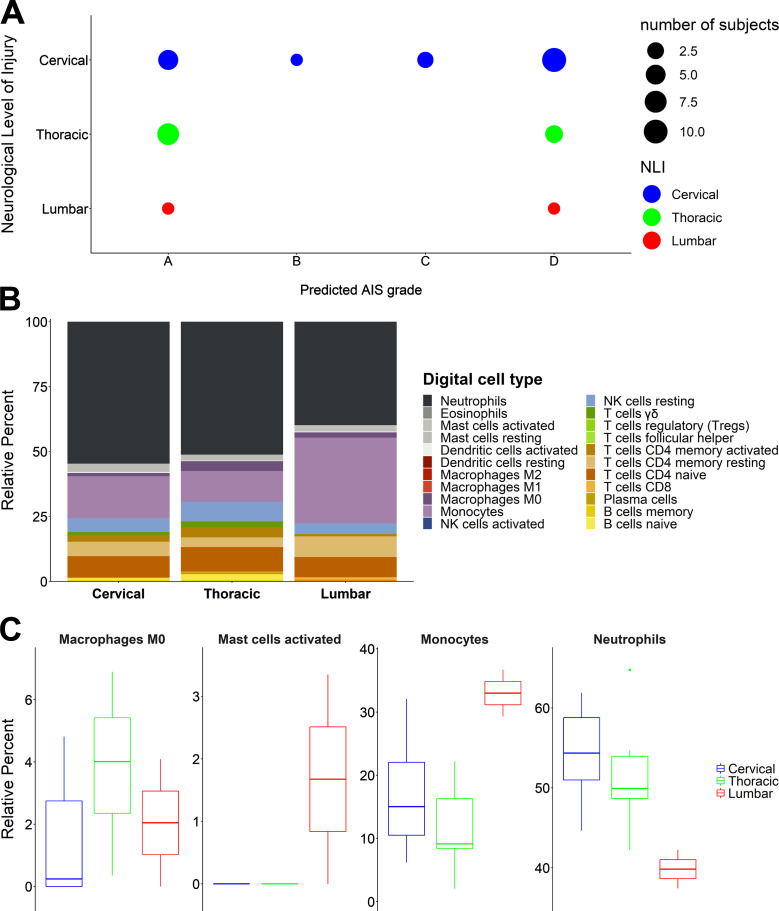
Diagnosis of spinal cord injury (SCI) severity at the ultra-acute stage is of great importance for emergency clinical care of patients as well as for potential enrollment into clinical trials. The lack of a diagnostic biomarker for SCI has played a major role in the poor results of clinical trials. We analyzed global gene expression in peripheral white blood cells during the acute injury phase and identified 197 genes whose expression changed after SCI compared with healthy and trauma controls and in direct relation to SCI severity. Unsupervised coexpression network analysis identified several gene modules that predicted injury severity (AIS grades) with an overall accuracy of 72.7% and included signatures of immune cell subtypes. Specifically, for complete SCIs (AIS A), ROC analysis showed impressive specificity and sensitivity (AUC: 0.865). Similar precision was also shown for AIS D SCIs (AUC: 0.938). Our findings indicate that global transcriptomic changes in peripheral blood cells have diagnostic and potentially prognostic value for SCI severity.
© 2021 Kyritsis et al.
Conflict of interest statement
Disclosures: G.T. Manley reported grants from NIH-NINDS and grants from United States Department of Defense during the conduct of the study. S. Dhall reported personal fees from Depuy Synthes, personal fees from Globus Medical, and other from Great Circle Technologies outside the submitted work. M.S. Beattie reported that he is on the board of directors of two nonprofits that support spinal cord injury research, the American Spinal Injury Association and the Praxis Spinal Cord Institute. Only travel expenses are reimbursed. No other disclosures were reported.
Figures

References
-
- Adler, D., and Murdoch D.. 2019. rgl: 3D Visualization Using OpenGL. R package version 0.100.30. https://CRAN.R-project.org/package=rgl
-
- Andrews, S. 2010. FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc
-
- Biering-Sørensen, F., Alai S., Anderson K., Charlifue S., Chen Y., DeVivo M., Flanders A.E., Jones L., Kleitman N., Lans A., et al. . 2015. Common data elements for spinal cord injury clinical research: a National Institute for Neurological Disorders and Stroke project. Spinal Cord. 53:265–277. 10.1038/sc.2014.246 - DOI - PMC - PubMed
-
- Blighe, K. 2019a. EnhancedVolcano: Publication-ready volcano plots with enhanced colouring and labeling. R package version 1.2.0. https://github.com/kevinblighe/EnhancedVolcano
-
- Blighe, K. 2019b. PCAtools: PCAtools: Everything Principal Components Analysis. R package version 1.1.10. https://github.com/kevinblighe/PCAtools
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
