

Targeting the AnxA1/Fpr2/ALX pathway regulates neutrophil function, promoting thromboinflammation resolution in sickle cell disease
- PMID: 33512489
- PMCID: PMC7976506
- DOI: 10.1182/blood.2020009166
Targeting the AnxA1/Fpr2/ALX pathway regulates neutrophil function, promoting thromboinflammation resolution in sickle cell disease
Abstract
Neutrophils play a crucial role in the intertwined processes of thrombosis and inflammation. An altered neutrophil phenotype may contribute to inadequate resolution, which is known to be a major pathophysiological contributor of thromboinflammatory conditions such as sickle cell disease (SCD). The endogenous protein annexin A1 (AnxA1) facilitates inflammation resolution via formyl peptide receptors (FPRs). We sought to comprehensively elucidate the functional significance of targeting the neutrophil-dependent AnxA1/FPR2/ALX pathway in SCD. Administration of AnxA1 mimetic peptide AnxA1Ac2-26 ameliorated cerebral thrombotic responses in Sickle transgenic mice via regulation of the FPR2/ALX (a fundamental receptor involved in resolution) pathway. We found direct evidence that neutrophils with SCD phenotype play a key role in contributing to thromboinflammation. In addition, AnxA1Ac2-26 regulated activated SCD neutrophils through protein kinase B (Akt) and extracellular signal-regulated kinases (ERK1/2) to enable resolution. We present compelling conceptual evidence that targeting the AnxA1/FPR2/ALX pathway may provide new therapeutic possibilities against thromboinflammatory conditions such as SCD.
© 2021 by The American Society of Hematology.
Conflict of interest statement
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Figures
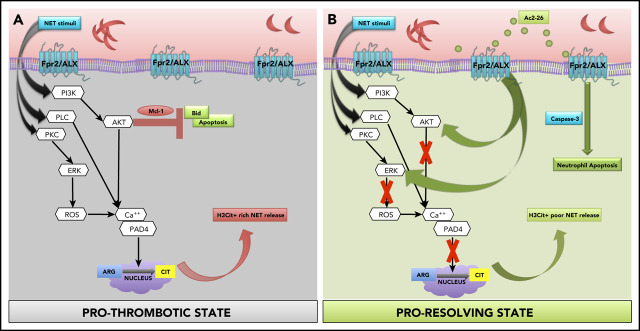
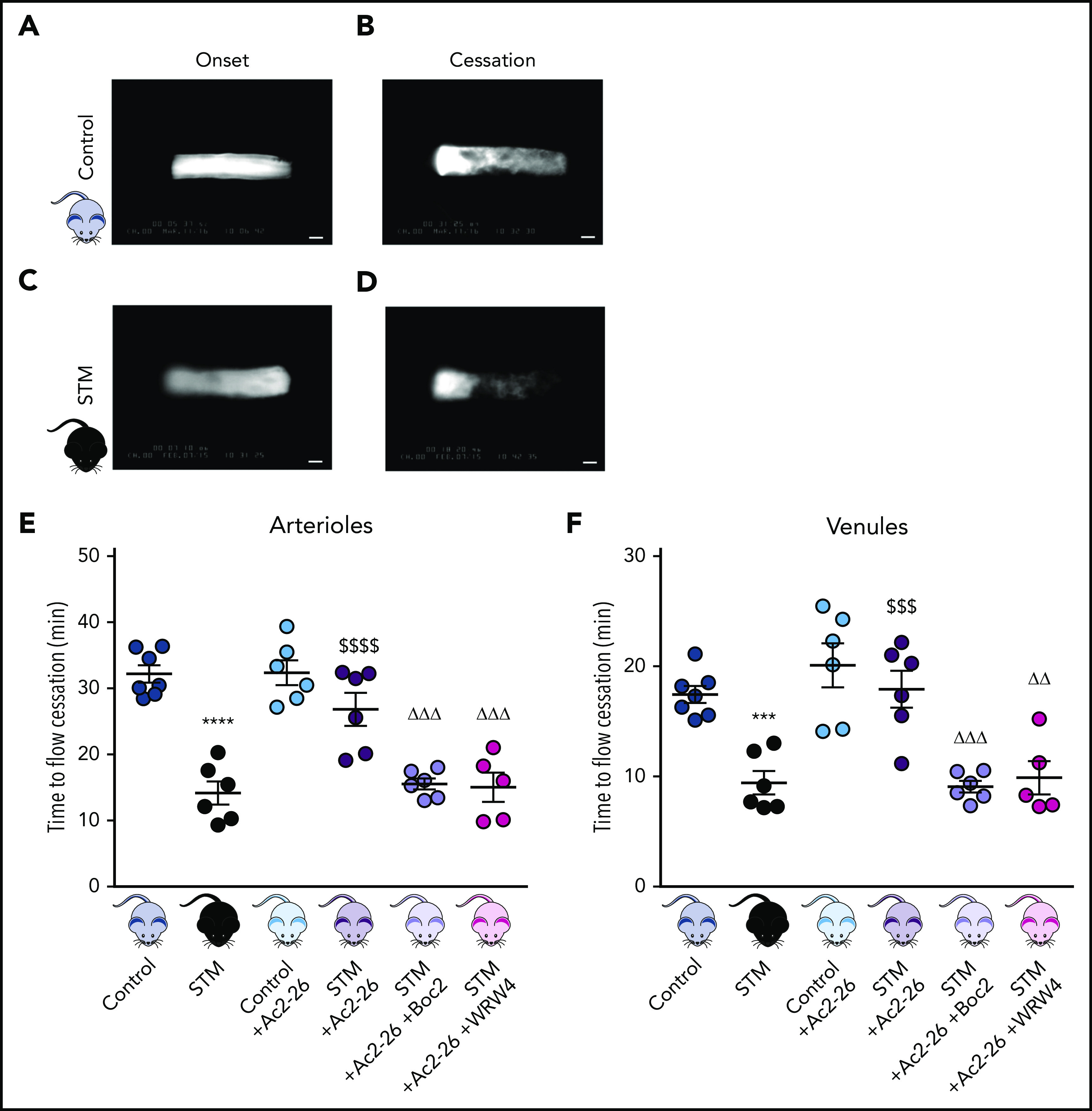
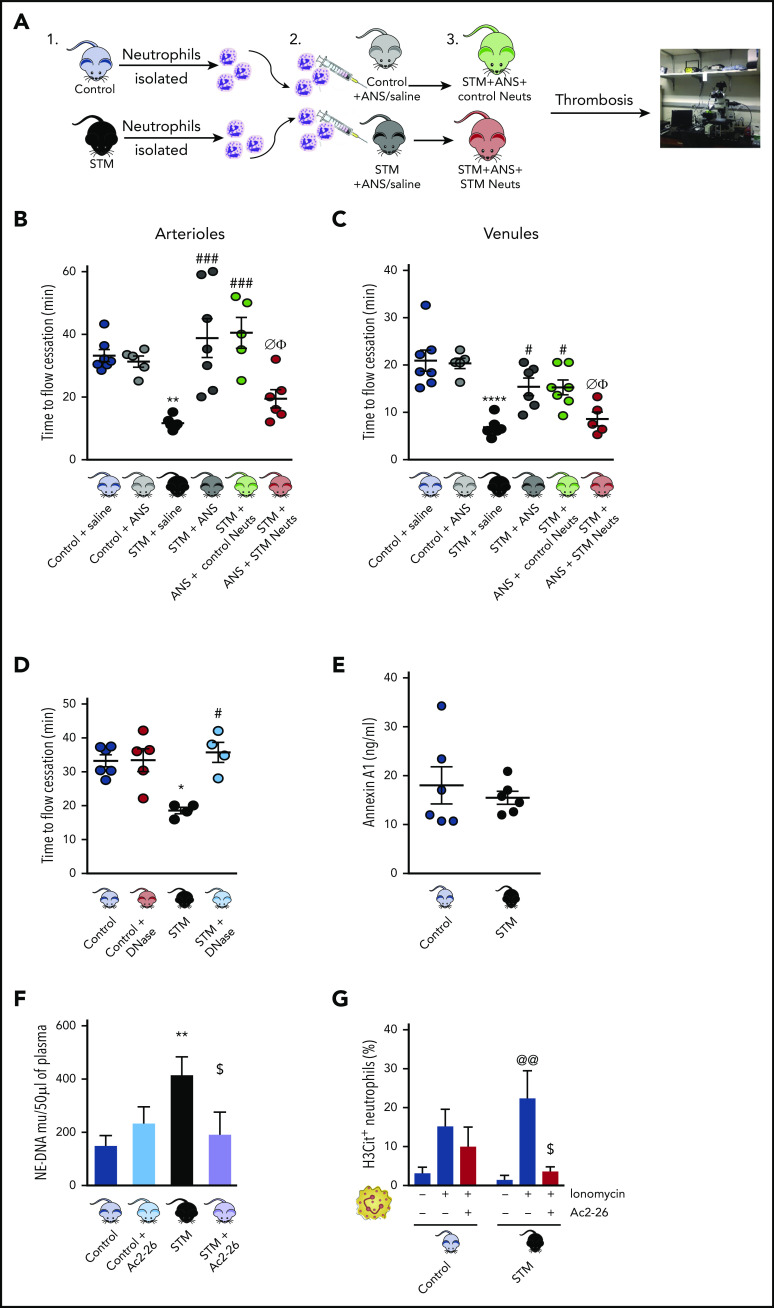
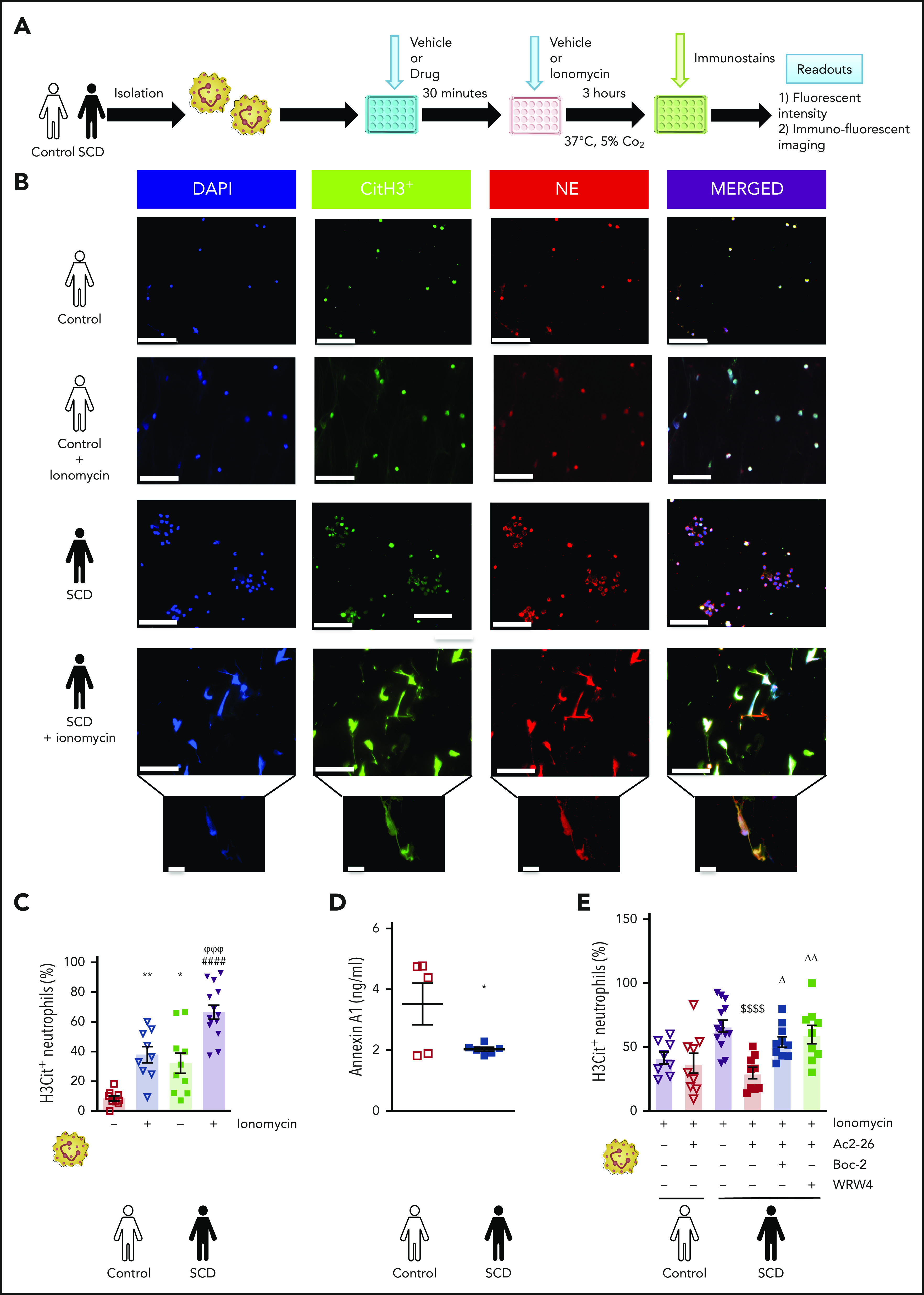
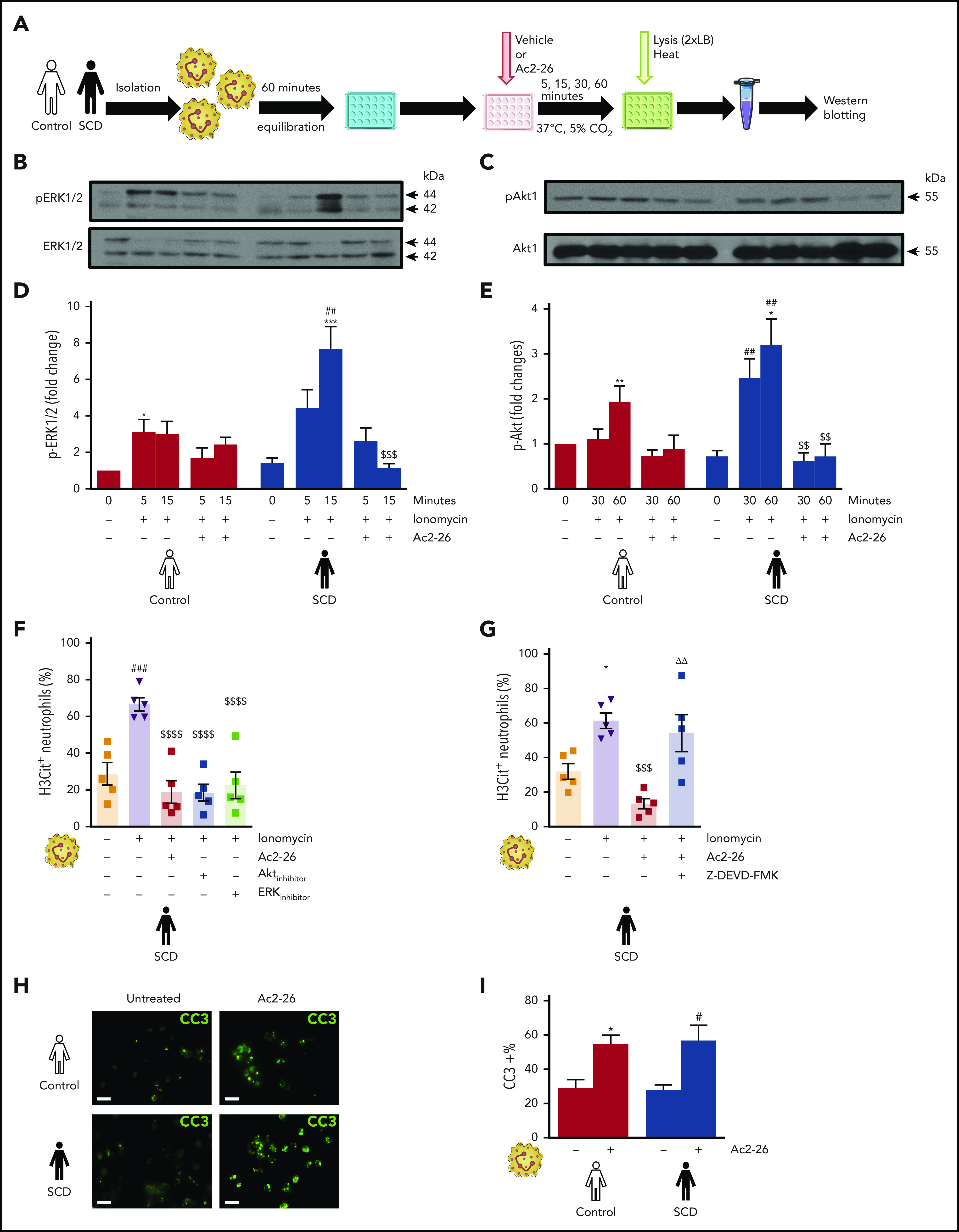
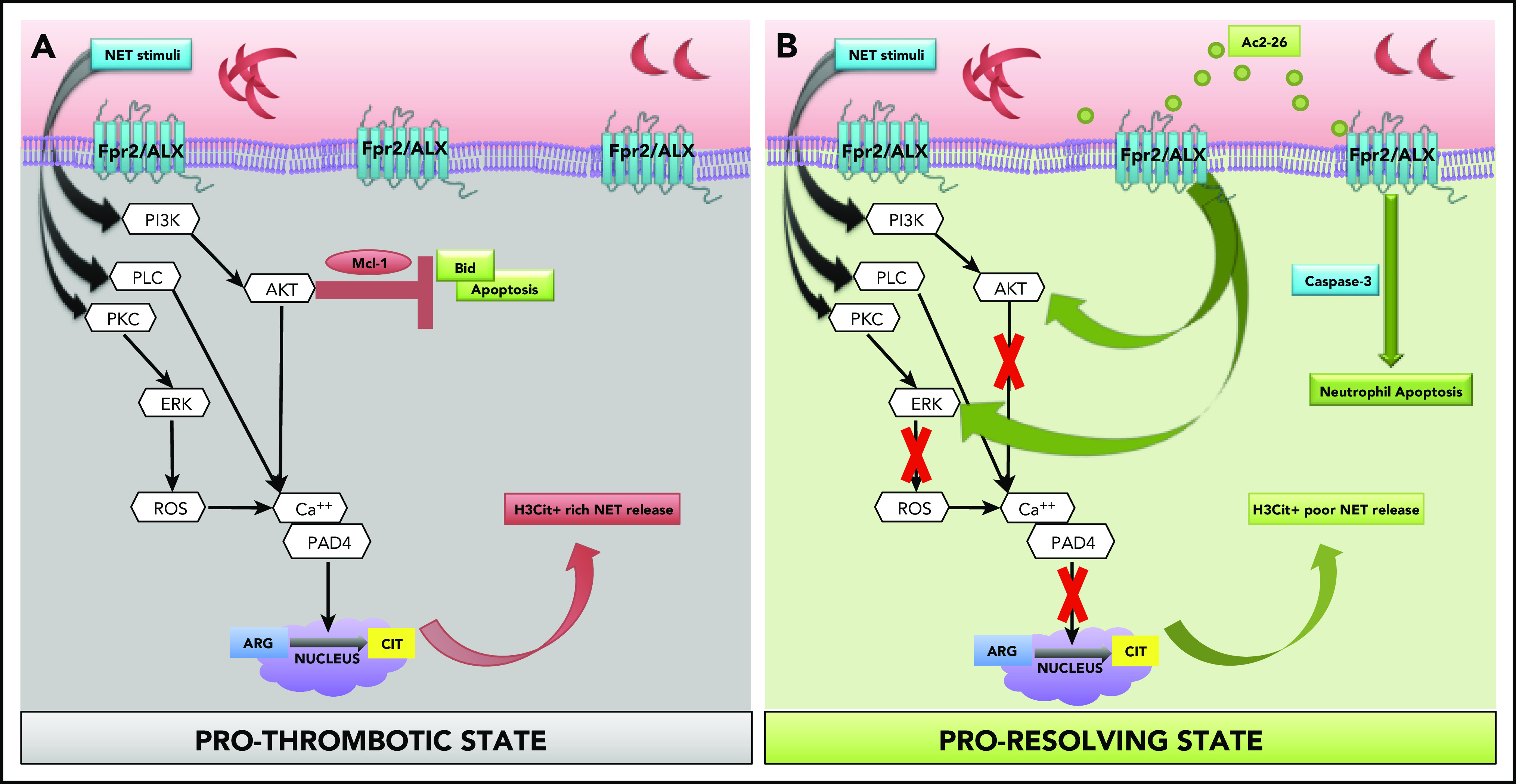
Comment in
-
Resolving thromboinflammation.Blood. 2021 Mar 18;137(11):1444-1446. doi: 10.1182/blood.2020010627. Blood. 2021. PMID: 33734338 No abstract available.
References
-
- Jackson SP, Darbousset R, Schoenwaelder SM. Thromboinflammation: challenges of therapeutically targeting coagulation and other host defense mechanisms. Blood. 2019;133(9):906-918. - PubMed
-
- Esmon CT. Crosstalk between inflammation and thrombosis. Maturitas. 2004;47(4):305-314. - PubMed
-
- Hess K, Grant PJ. Inflammation and thrombosis in diabetes. Thromb Haemost. 2011;105(S 06 suppl 1):S43-S54. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
