

Neurotoxicity and underlying cellular changes of 21 mitochondrial respiratory chain inhibitors
- PMID: 33512557
- PMCID: PMC7870626
- DOI: 10.1007/s00204-020-02970-5
Neurotoxicity and underlying cellular changes of 21 mitochondrial respiratory chain inhibitors
Abstract
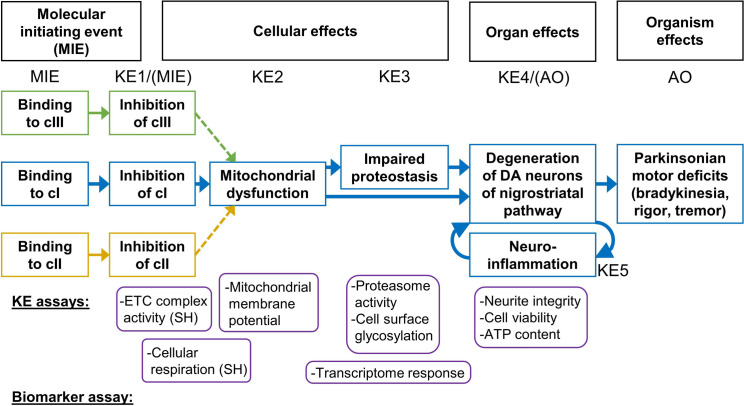
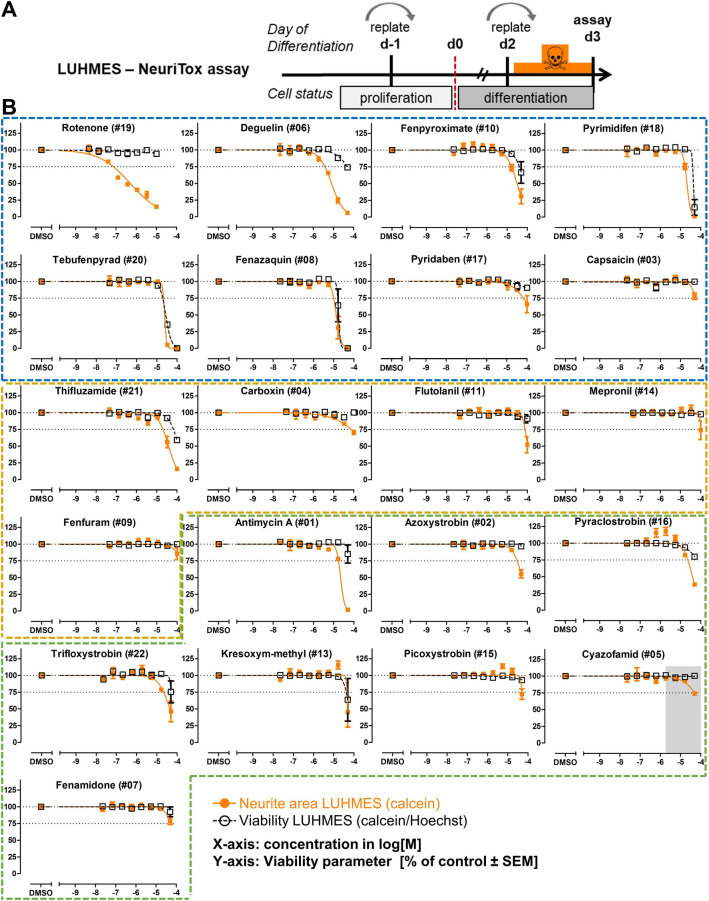
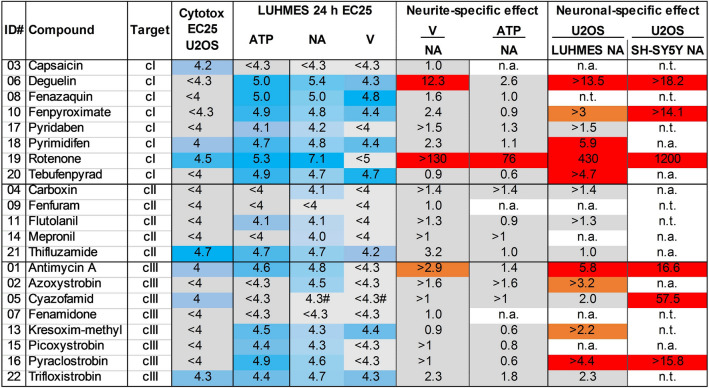
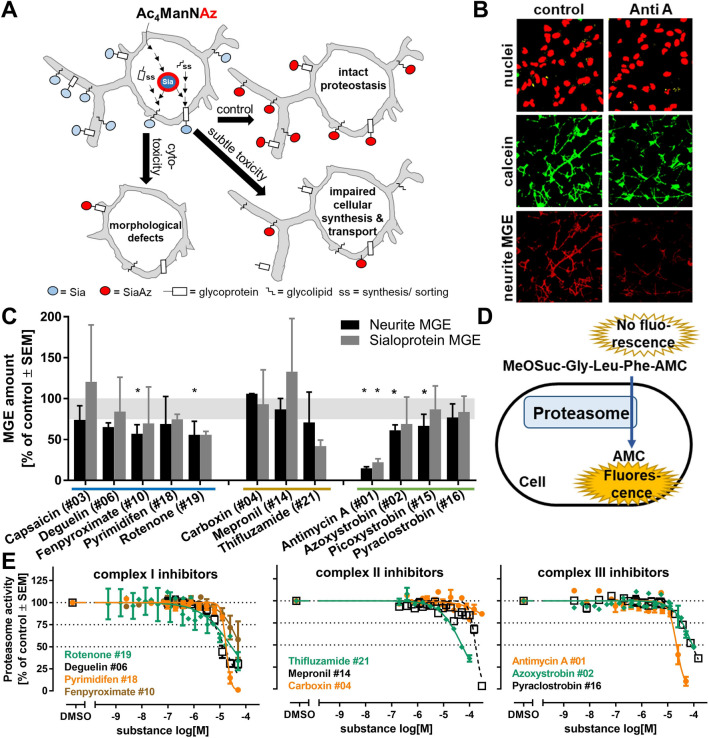
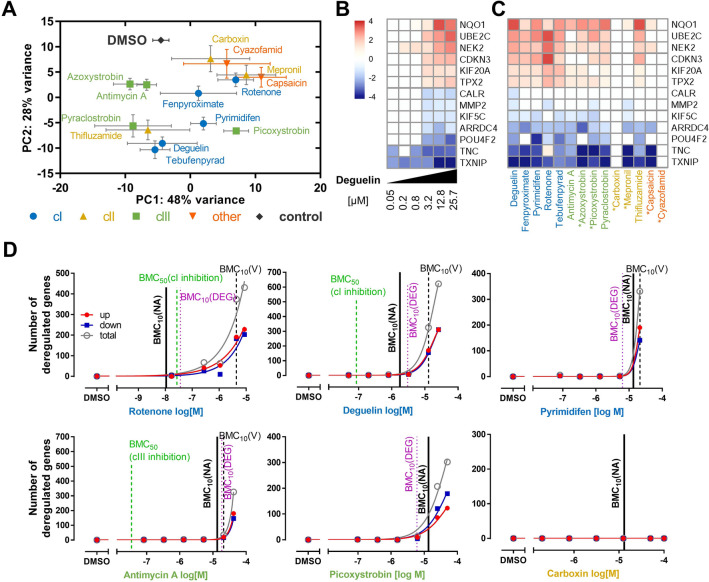
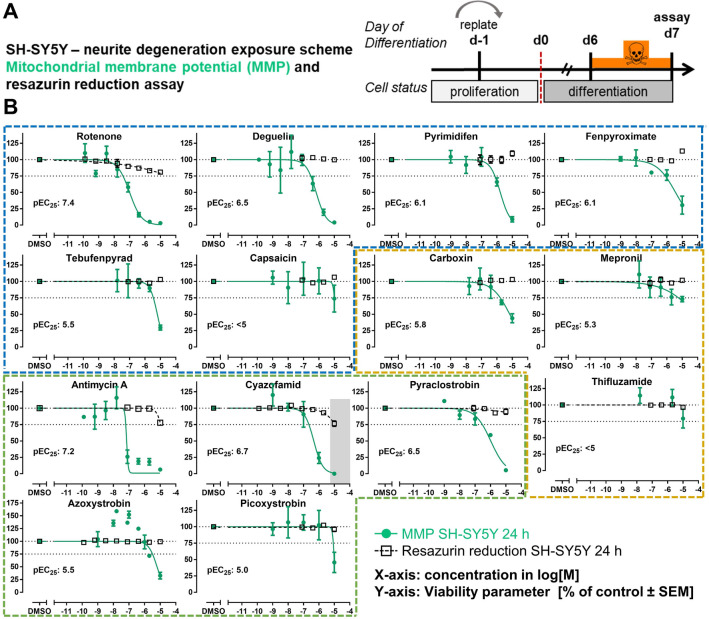
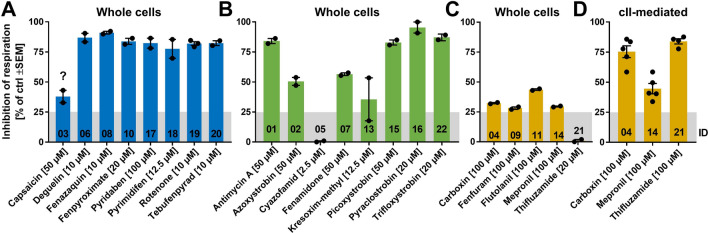
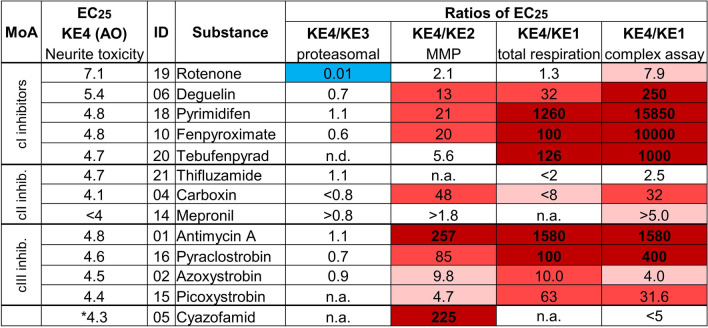
Inhibition of complex I of the mitochondrial respiratory chain (cI) by rotenone and methyl-phenylpyridinium (MPP +) leads to the degeneration of dopaminergic neurons in man and rodents. To formally describe this mechanism of toxicity, an adverse outcome pathway (AOP:3) has been developed that implies that any inhibitor of cI, or possibly of other parts of the respiratory chain, would have the potential to trigger parkinsonian motor deficits. We used here 21 pesticides, all of which are described in the literature as mitochondrial inhibitors, to study the general applicability of AOP:3 or of in vitro assays that are assessing its activation. Five cI, three complex II (cII), and five complex III (cIII) inhibitors were characterized in detail in human dopaminergic neuronal cell cultures. The NeuriTox assay, examining neurite damage in LUHMES cells, was used as in vitro proxy of the adverse outcome (AO), i.e., of dopaminergic neurodegeneration. This test provided data on whether test compounds were unspecific cytotoxicants or specifically neurotoxic, and it yielded potency data with respect to neurite degeneration. The pesticide panel was also examined in assays for the sequential key events (KE) leading to the AO, i.e., mitochondrial respiratory chain inhibition, mitochondrial dysfunction, and disturbed proteostasis. Data from KE assays were compared to the NeuriTox data (AO). The cII-inhibitory pesticides tested here did not appear to trigger the AOP:3 at all. Some of the cI/cIII inhibitors showed a consistent AOP activation response in all assays, while others did not. In general, there was a clear hierarchy of assay sensitivity: changes of gene expression (biomarker of neuronal stress) correlated well with NeuriTox data; mitochondrial failure (measured both by a mitochondrial membrane potential-sensitive dye and a respirometric assay) was about 10-260 times more sensitive than neurite damage (AO); cI/cIII activity was sometimes affected at > 1000 times lower concentrations than the neurites. These data suggest that the use of AOP:3 for hazard assessment has a number of caveats: (i) specific parkinsonian neurodegeneration cannot be easily predicted from assays of mitochondrial dysfunction; (ii) deriving a point-of-departure for risk assessment from early KE assays may overestimate toxicant potency.
Keywords: AOP:3; High-content imaging; In vitro neurotoxicity; Mechanistic safety assessment; Mitotoxicity; TempO-Seq.
Conflict of interest statement
The authors declare that there is no conflict of interest.
Figures

Similar articles
-
Development of a neurotoxicity assay that is tuned to detect mitochondrial toxicants.Arch Toxicol. 2019 Jun;93(6):1585-1608. doi: 10.1007/s00204-019-02473-y. Epub 2019 Jun 12. Arch Toxicol. 2019. PMID: 31190196
-
An adverse outcome pathway for parkinsonian motor deficits associated with mitochondrial complex I inhibition.Arch Toxicol. 2018 Jan;92(1):41-82. doi: 10.1007/s00204-017-2133-4. Epub 2017 Dec 5. Arch Toxicol. 2018. PMID: 29209747 Free PMC article. Review.
-
Rotenone Increases Isoniazid Toxicity but Does Not Cause Significant Liver Injury: Implications for the Hypothesis that Inhibition of the Mitochondrial Electron Transport Chain Is a Common Mechanism of Idiosyncratic Drug-Induced Liver Injury.Chem Res Toxicol. 2019 Jul 15;32(7):1423-1431. doi: 10.1021/acs.chemrestox.9b00116. Epub 2019 Jun 28. Chem Res Toxicol. 2019. PMID: 31251588
-
Reactive oxygen species are generated by the respiratory complex II--evidence for lack of contribution of the reverse electron flow in complex I.FEBS J. 2013 Feb;280(3):927-38. doi: 10.1111/febs.12086. Epub 2013 Jan 7. FEBS J. 2013. PMID: 23206332
-
New inhibitors of complex I of the mitochondrial electron transport chain with activity as pesticides.Biochem Soc Trans. 1994 Feb;22(1):230-3. doi: 10.1042/bst0220230. Biochem Soc Trans. 1994. PMID: 8206238 Review. No abstract available.
Cited by
-
Definition of the Neurotoxicity-Associated Metabolic Signature Triggered by Berberine and Other Respiratory Chain Inhibitors.Antioxidants (Basel). 2023 Dec 28;13(1):49. doi: 10.3390/antiox13010049. Antioxidants (Basel). 2023. PMID: 38247474 Free PMC article.
-
New approach methods to assess developmental and adult neurotoxicity for regulatory use: a PARC work package 5 project.Front Toxicol. 2024 Apr 26;6:1359507. doi: 10.3389/ftox.2024.1359507. eCollection 2024. Front Toxicol. 2024. PMID: 38742231 Free PMC article.
-
Recent advances and current challenges of new approach methodologies in developmental and adult neurotoxicity testing.Arch Toxicol. 2024 May;98(5):1271-1295. doi: 10.1007/s00204-024-03703-8. Epub 2024 Mar 13. Arch Toxicol. 2024. PMID: 38480536 Free PMC article. Review.
-
Assessing the Neurodevelopmental Impact of Fluoxetine, Citalopram, and Paroxetine on Neural Stem Cell-Derived Neurons.Pharmaceuticals (Basel). 2024 Oct 18;17(10):1392. doi: 10.3390/ph17101392. Pharmaceuticals (Basel). 2024. PMID: 39459031 Free PMC article.
-
The Potential of Edible and Medicinal Resource Polysaccharides for Prevention and Treatment of Neurodegenerative Diseases.Biomolecules. 2023 May 22;13(5):873. doi: 10.3390/biom13050873. Biomolecules. 2023. PMID: 37238743 Free PMC article. Review.
References
-
- Bal-Price A, Leist M, Schildknecht S, Tschudi-Monnet F, Paini A, Terron A (2018) Adverse outcome pathway on inhibition of the mitochondrial complex I of nigro-striatal neurons leading to parkinsonian motor deficits. OECD Publishing, Paris. 10.1787/b46c3c00-en
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
