

Graph Algorithms for Mixture Interpretation
- PMID: 33514030
- PMCID: PMC7911948
- DOI: 10.3390/genes12020185
Graph Algorithms for Mixture Interpretation
Abstract
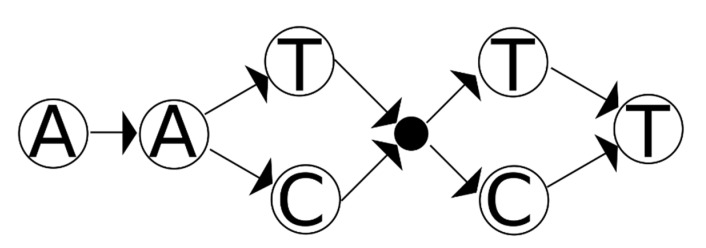
The scale of genetic methods are presently being expanded: forensic genetic assays previously were limited to tens of loci, but now technologies allow for a transition to forensic genomic approaches that assess thousands to millions of loci. However, there are subtle distinctions between genetic assays and their genomic counterparts (especially in the context of forensics). For instance, forensic genetic approaches tend to describe a locus as a haplotype, be it a microhaplotype or a short tandem repeat with its accompanying flanking information. In contrast, genomic assays tend to provide not haplotypes but sequence variants or differences, variants which in turn describe how the alleles apparently differ from the reference sequence. By the given construction, mitochondrial genetic assays can be thought of as genomic as they often describe genetic differences in a similar way. The mitochondrial genetics literature makes clear that sequence differences, unlike the haplotypes they encode, are not comparable to each other. Different alignment algorithms and different variant calling conventions may cause the same haplotype to be encoded in multiple ways. This ambiguity can affect evidence and reference profile comparisons as well as how "match" statistics are computed. In this study, a graph algorithm is described (and implemented in the MMDIT (Mitochondrial Mixture Database and Interpretation Tool) R package) that permits the assessment of forensic match statistics on mitochondrial DNA mixtures in a way that is invariant to both the variant calling conventions followed and the alignment parameters considered. The algorithm described, given a few modest constraints, can be used to compute the "random man not excluded" statistic or the likelihood ratio. The performance of the approach is assessed in in silico mitochondrial DNA mixtures.
Keywords: graph algorithm; massively parallel sequencing; mitochondrial mixtures; mixture interpretation; probabilistic genotyping.
Conflict of interest statement
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Figures










References
-
- Krawczak M. Forensic interpretation of haploid DNA mixtures. Int. Congress Ser. 2006;1288:477–483. doi: 10.1016/j.ics.2005.10.041. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
